Dyrektor Centrum Rozwoju Badań Klinicznych w Agencji Badań Medycznych
Każdy lek, który kupujemy w aptece, przeszedł wcześniej fazy ściśle kontrolowanych badań klinicznych. W czasie czterech faz testuje się jego bezpieczeństwo, dawkowanie, reakcje z innymi schorzeniami lub lekami. Monitorowanie nie kończy się z momentem wypuszczenia leku na rynek. – Leki mają serie. Każda seria leku musi być taka sama, to znaczy skład musi być taki sam – tłumaczy dr n. med. Elżbieta Bylina, dyrektorka Centrum Rozwoju Badań Klinicznych w Agencji Badań Medycznych. Monitoruje się też zgłoszenia od pacjentów i lekarzy na temat ewentualnych działań niepożądanych i jeśli jest ich dużo, lek może zostać wycofany z rynku. Te przepisy nie dotyczą suplementów diety: nie muszą przechodzić badań klinicznych ani zachowywać niezmiennego składu. Suplementy diety to żywność, nie mogą leczyć, choć mogą działać wspierająco. Pamiętajmy o tym widząc kolejne reklamy suplementów.
Nad wprowadzaniem na rynek nowych leków czuwają instytucje państwowe. W Polsce są to Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych oraz Naczelna Komisja Bioetyczna. Dodatkowe komisje bioetyczne można powoływać przy uniwersytetach, placówkach badawczych i izbach lekarskich.
Działanie testowanej substancji leczniczej sprawdza się najpierw na sztucznie wyhodowanych komórkach, ale to za mało. Wszystkie leki są na jakimś etapie testowane na zwierzętach i na pacjentach, którzy wyrazili na to zgodę. – Dana cząsteczka może się inaczej zachowywać wewnątrz organizmu niż na zewnątrz, na liniach komórkowych – tłumaczy dr Bylina. Naukowcy starają się zwierzęta oszczędzać, np. testować kilka różnych substancji na tym samym osobniku. A badania na ludziach są dobrowolne (ochotnicy zgłaszają się do badań klinicznych, ważne: nigdy za to nie płacą) i zwykle podwójnie zaślepione. To znaczy, że pacjent nie wie, czy otrzymuje badany produkt, czy placebo, nie wie tego również lekarz. To zapewnia obiektywność wyników. Naukowcy starają się też, by grupy badawcze były zróżnicowane pod względem płci, wieku i chorób towarzyszących. To pozwala lepiej przetestować produkt i wyłapać ewentualne skutki uboczne na wcześniejszym etapie.
W odcinku usłyszycie też, dlaczego kobiety w ciąży i małe dzieci mogą przyjmować tak mało leków, gdzie szukać informacji o badaniach klinicznych i jak zapewnić, by producent leku nie wpływał na wyniki badań. Są też słowa podziękowania dla wszystkich pacjentów, którzy decydują się brać udział w badaniach.
Odcinek jest efektem współpracy z Agencją Badań Medycznych, publicznej instytucji działającej od 2019 roku.
TRANSKRYPCJA
Karolina Głowacka: W studio Radia Naukowego pani doktor nauk medycznych Elżbieta Bylina. Dzień dobry.
Elżbieta Bylina: Dzień dobry.
K.G.: Z Agencji Badań Medycznych. Pani doktor jest dyrektorką Centrum Rozwoju Badań Klinicznych w ABM właśnie, a będziemy rozmawiały rzecz jasna o badaniach klinicznych. Bardzo ciekawy temat. Temat jednocześnie złożony, temat, o którym może nie myślimy na co dzień, ale efekty tychże badań klinicznych dotyczą nas masowo jako pacjentów i pacjentki. Pani doktor, czy to jest tak, że każdy lek, który trafił do aptek czy do zastosowania szpitalnego, musiał przejść taką drogę badań klinicznych?
E.B.: Tak, musiał. Jeżeli mówimy o lekach, to każdy lek, czyli substancja, którą nazywamy lekiem, musi przejść przez badania kliniczne.
K.G.: I to się różni, o ile się orientuję, od suplementów.
E.B.: Tak. Ta różnica jest fundamentalna, dlatego że należy pamiętać, że wszystkie leki muszą udowodnić skuteczność i bezpieczeństwo, w przeciwieństwie do suplementów, które nie muszą przechodzić drogi badań klinicznych. I oczywiście każdy lek, każda seria leku, bo wiadomo, że leki mają serie, musi być taka sama, to znaczy ten skład musi być taki sam. Natomiast jeżeli chodzi o suplementy, między seriami ten skład może być inny. I tak się zdarza. Ja wiem, że czasami te reklamy suplementów są takie, że możemy odnieść wrażenie, że to są leki, ale to nie są leki. Nie wymagają badań klinicznych, podkreślam, i nie musimy udowodniać ich skuteczności.
K.G.: Ale wyglądają tak podobnie. Są w blistrach, w tabletkach, są w aptekach przecież.
E.B.: Tak, bardzo. Tak. Jednak pamiętajmy o tym, że suplementy nie są lekami. Suplementy tak naprawdę możemy traktować jako żywność.
K.G.: I teoretycznie gdzieś tam ta informacja się przebija, ale wydaje mi się, że bardzo powoli. Swoją drogą nie wiem, czy to nie jest jakiś pomysł na poważniejsze regulacje, żeby to na przykład było wyraźnie zaznaczone w aptekach, nie?
E.B.: Tak, tak, to powinno być jasno zaznaczone, dlatego że oczywiście osoby, które interesują się badaniami klinicznymi, są profesjonalistami, czy to farmaceuci, czy pracownicy ochrony zdrowia, są na to wyczuleni, mają wiedzę, natomiast oczywiście przeciętna osoba, która wchodzi do apteki, może mieć z tym problem i absolutnie ja też uważam, że takie bardzo wyraźne oznaczenie, co jest lekiem, a co jest suplementem, byłoby wskazane.
K.G.: Ja to bym też była za tym, żeby tak zwanych leków homeopatycznych, które nie mogą działać, ponieważ po prostu dawka jest tam tak rozcieńczona, że fizycznie nie są w stanie działać na nasz organizm, żeby też nie było w aptekach.
E.B.: Tak. Tak, tutaj są… Tak. Musimy dbać o pewną transparentność, przejrzystość i czystość pewnych informacji.
K.G.: To tyle, jeśli chodzi o apteki, a teraz właśnie same badania kliniczne. Kiedy w ogóle się stało coś takiego, że medycyna wpadła na taki pomysł, żeby testować leki właśnie w sposób taki standaryzowany, proceduralny, dokładny? Kiedy to się stało?
E.B.: To się tak naprawdę stało w latach 50., 60. XX wieku. Tak naprawdę te regulacje, z którymi mamy w tej chwili do czynienia, które w tej chwili regulują cały proces badania klinicznego, one zostały przygotowane po tej tragedii talidomidu w latach, tak jak powiedziałam, 50., 60. ubiegłego wieku. I to wydarzenie doprowadziło do bardzo dużego zaostrzenia przepisów i stworzenia tak naprawdę dzisiejszych standardów badań klinicznych. Tak że to był ten moment.
K.G.: A talidomid to był ten lek na nudności? Dobrze kojarzę?
E.B.: Tak, tak. Talidomid tak naprawdę to był lek uspokajający, ale generalnie chodziło o to, żeby powstrzymywał wymioty u kobiet ciężarnych. I wiadomo, że oczywiście te wymioty najczęściej są w pierwszym trymestrze ciąży, i on był nagminnie przepisywany kobietom ciężarnym. Wiadomo, co on robił. Oczywiście powodował uszkodzenia płodu w postaci tego, że u płodów nie wykształcały się kończyny. No i oczywiście odkryto to dopiero wtedy, kiedy te dzieci się urodziły i oczywiście przeanalizowano przypadki właśnie takich urodzeń i zaczęto szukać, z jakiego powodu to się wydarzyło. No i wtedy okazało się, że w zasadzie u wszystkich tych kobiet, wszystkie te kobiety, które urodziły dzieci z tak zwaną fokalią, czyli brakiem kończyn, właśnie one zażywały talidomid.
K.G.: Ale to się położyło cieniem w ogóle na rynek leków, tak myślę.
E.B.: Tak.
K.G.: A pani mi mówi, że to była ta sytuacja, która spowodowała właśnie zaostrzenie procedur.
E.B.: Tak, dlatego że właśnie po tej sytuacji… To się położyło cieniem. Oczywiście, że się położyło cieniem.
K.G.: I się do tej pory przypomina przecież, wspomina.
E.B.: Tak, przypomina się. Natomiast ta sytuacja wymogła chociażby to, żeby podczas badania klinicznego badać wpływ danej substancji, która może być potencjalnym lekiem, na płód. I to są takie kamienie milowe, bo z jednej strony mamy te nadużycia w badaniach klinicznych, oczywiście historia tych nadużyć jest bardzo długa, ale te nadużycia, my wyciągamy z nich wnioski, czyli później po prostu staramy się zapobiegać tym sytuacjom, które się wydarzyły.
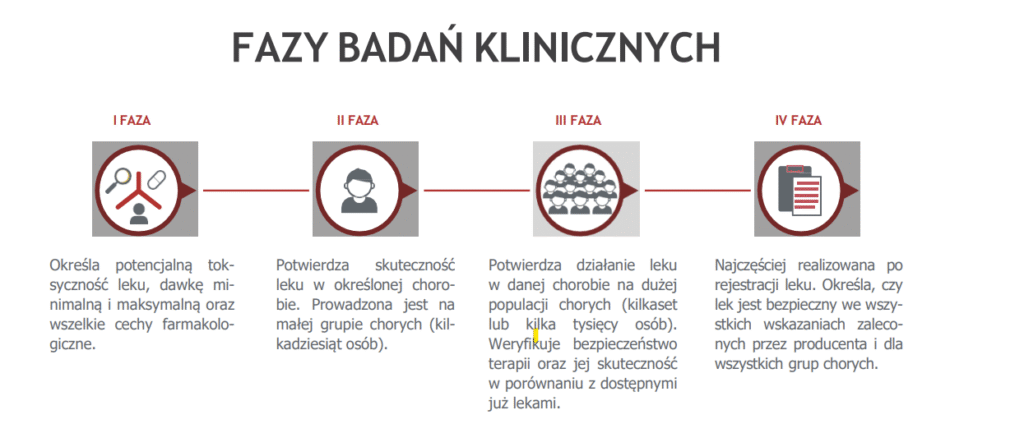
K.G.: Jakie są etapy badań klinicznych? Cztery fazy są, o ile wiem.
E.B.: Tak. Mamy cztery fazy badań klinicznych. Pierwsza faza to jest oczywiście first in human, pierwsze podanie u człowieka. I to jest pierwsza faza, która ma potwierdzić przede wszystkim bezpieczeństwo.
K.G.: Ale to w takim razie skoro ta pierwsza faza już jest pierwszym momentem, kiedy substancje podaje się człowiekowi, no to mamy jeszcze, to może powiedzmy, fazy badań przedklinicznych.
E.B.: Tak, oczywiście. Mamy fazy badań przedklinicznych. Tak naprawdę ten proces jest bardzo długi i bardzo złożony. Więc zaczynamy tak naprawdę w laboratorium. Naukowcy najczęściej…
K.G.: Od pomysłu.
E.B.: Tak, od pomysłu. Czyli mamy pewną ideę, pomysł. I najbardziej obrazowo to wygląda tak, że naukowcy identyfikują powiedzmy jakiś cel biologiczny, na przykład białko, które powoduje jakąś chorobę. I wtedy mamy zidentyfikowany cel i wtedy staramy się znaleźć substancję, cząsteczkę, która w jakiś sposób będzie wpływać na ten nasz, ogólnie mówiąc, cel. Czyli na to… Będzie zmieniać w jakiś sposób to białko, powiedzmy. I w laboratorium analizuje się naprawdę dziesiątki tysięcy cząsteczek. Oczywiście tak naprawdę większość, powyżej 90% tych cząsteczek, po prostu nie przechodzi do dalszych badań. Później mówimy o badaniach przedklinicznych, czyli badaniach na modelach komórkowych, później badaniach na zwierzętach i dopiero w momencie, kiedy te wyniki są na tyle optymistyczne, to znaczy na tyle bezpieczne i widzimy pewien potencjał skuteczności danej cząsteczki, to wtedy zaczynamy prowadzić badania z udziałem ludzi i to są właśnie badania kliniczne. Jeżeli mówimy o badaniach z udziałem ludzi, to oczywiście mamy, tak jak pani słusznie zauważyła, mamy cztery fazy badań. Pierwsza to jest oczywiście badanie pierwszej fazy. W czasie tej fazy badamy przede wszystkim bezpieczeństwo i możemy tutaj już oceniać dawkę, czyli oceniamy, jaka dawka będzie jednocześnie bezpieczna i skuteczna. Druga faza to jest głównie po to, aby potwierdzić skuteczność. I tutaj w pierwszej bezpieczeństwo, czyli mamy lek bezpieczny. Druga faza: oceniamy skuteczność, czyli osiągamy ten założony cel, czyli cząsteczka jest skuteczna. I trzecia faza to jest: porównujemy, czy ta nowa cząsteczka jest lepsza od standardowego leczenia, jeżeli oczywiście takie mamy. Ta trzecia faza też jest nazywana fazą rejestracyjną, dlatego że po zakończeniu trzeciej fazy badania klinicznego możemy złożyć wniosek do odpowiednich urzędów o to, żeby ta cząsteczka została zarejestrowana. I wprowadzamy cząsteczkę do obrotu i wtedy już możemy nazywać ją lekiem. I mamy jeszcze czwartą fazę, fazę porejestracyjną, gdzie badamy już lek na szerszej populacji, w takim ogólnym dostępie. Tak że takie są etapy. Tak jak powiedziałam, droga jest bardzo długa i bardzo złożona. Ten cały proces od pomysłu do rejestracji może trwać między 10 a 15 lat. Także to jest naprawdę bardzo długi proces i bardzo, bardzo złożony.
K.G.: Czy w takim razie z tej czwartej fazy biorą się te informacje, które czytamy w ulotkach, że tam jeśli weźmiesz ten lek na ból głowy, to u jednej na tysiąc osób może się zdarzyć cośtam, na przykład nudności.
E.B.: To jest trzecia faza, czyli po trzeciej fazie, bo to mamy w tak zwanej charakterystyce produktu leczniczego. I ta charakterystyka powstaje wtedy, kiedy lek jest rejestrowany. I sponsor, czyli tak naprawdę już producent tego leku, musi przygotować właśnie charakterystykę. I po prostu na podstawie badań fazy trzeciej właśnie my szacujemy, jakie zdarzenie niepożądane czy działanie niepożądane tego leku, jak często ono występuje, prawda? Że właśnie tak jak pani powiedziała, że u jednej na dziesięć osób albo u jednej na tysiąc osób wystąpi takie czy inne działania niepożądane.
K.G.: Tak jeszcze wracam myślami do tego etapu badanie przedklinicznych, czyli testowania na zwierzętach. Czy nie może być tak, że część leków, które mogą zadziałać u ludzi, odpada na tym etapie? Bo przecież są takie substancje, które dla niektórych zwierząt są toksyczne. Nie chcę teraz nikogo wprowadzać w błąd, ale wydaje mi się, że jeden z leków, który jest u ludzi popularnie używany na ból głowy, właśnie dla kotów na przykład jest toksyczny i nie może być stosowany. I tak się zastanawiam, czy tam właśnie na tym etapie niektóre rzeczy nie przepadają.
E.B.: Tak, ale o tym myśli się wcześniej, planuje się, prawda? Czyli zależnie od tego, z jaką cząsteczką mamy do czynienia, testujemy na określonym zwierzęciu, żeby stworzyć mechanizm jak najbardziej podobny do tego mechanizmu u człowieka. Czyli na przykład możemy, część leków badamy na myszach czy na szczurach, czy na świniach, ale na przykład część leków też badamy na człekokształtnych. Więc to wszystko zależy od tego, z jaką cząsteczką mamy do czynienia.
K.G.: Czy są jakieś widoki, żeby maksymalnie ograniczyć właśnie badania na zwierzętach?
E.B.: Są takie widoki. Proszę też pamiętać, że jeżeli mówimy o badaniach na zwierzętach, to też mamy komisje etyczne, które wydają zgodę na to, żeby badanie było przeprowadzone na zwierzętach. Więc to też nie jest tak, że to jest zupełnie nieuregulowane, że naukowcy po prostu badają, testują nowe cząsteczki zupełnie nieograniczenie na zwierzętach. Tak że tutaj ja nie jestem naukowcem, który pracuje sensu stricte w laboratorium, natomiast wiem, że oczywiście są różne sposoby na to, żeby ograniczyć te badania, więc jeżeli mówimy o badaniach w onkologii na przykład, to wiem, że jeżeli sprawdzamy daną cząsteczkę na myszach, prowadzimy te badania na myszach, to po prostu podajemy… Jedna mysz, mówiąc najbardziej obrazowo, ma kilka guzów, kilka zmian, tak żeby po prostu sprawdzić różne rzeczy. Więc są pewne mechanizmy, które mają spowodować, że ograniczamy liczbę zwierząt, które poddajemy tym badaniom.
K.G.: Ja muszę przyznać, że dla mnie to też jest ważny temat i rozmawiałam z różnymi naukowcami na ten temat, licząc na to, że może jest jakiś nie tyle magiczny, co technologiczny sposób na to, żeby całkiem zrezygnować z tych badań, ale niestety mówią, że całkiem od tego odejść się nie da, bo nie da się wytworzyć organizmu. To jest po prostu złożona…
E.B.: Tak, to jest złożona struktura. I tak jak pani wspomniała, też nie da się wszystkiego zrobić na modelach komórkowych tylko i wyłącznie. Dlatego że no po prostu organizm jest organizmem. To jest bardzo złożona i skomplikowana struktura. Dana cząsteczka może się inaczej zachowywać wewnątrz organizmu niż na zewnątrz, na liniach komórkowych, więc jest to proces, którego… Nie unikniemy tego etapu badania na zwierzętach póki co, myślę, że w najbliższej przyszłości.
K.G.: Gra się toczy o to, żeby jak najbardziej to minimalizować.
E.B.: Tak.
K.G.: Bo są też, i też w Radiu Naukowym o tym kiedyś rozmawialiśmy, o takich organoidach, akurat o tak zwanych minimózgach rozmawialiśmy, czyli tworzy się właśnie takie w hodowlach komórkowych no, nie organy, w sensie nie jest tak, że sobie leży taka wątroba w laboratorium, ale ileś tam komórek, które są podobne i tam można też część cząsteczek testować. No ja oczywiście kibicuję, żeby maksymalnie ograniczać badania na zwierzętach. Pani doktor też tutaj kiwa głową. Przejdźmy teraz do badań klinicznych, które właśnie odbywają się już z udziałem pacjentów. Jak się projektuje takie badanie kliniczne? Może zatrzymajmy się na początek na tej pierwszej fazie, bo ona się wydaje, przynajmniej dla mnie, taka najbardziej stresująca, no bo badamy bezpieczeństwo, czyli musimy po raz pierwszy daną substancję podać człowiekowi. Jak się taką osobę w ogóle rekrutuje? Jak to działa?
E.B.: Jeżeli chodzi o sam projekt badania, to mamy coś takiego jak protokół badania klinicznego, gdzie w tym właśnie protokole badania klinicznego wszystko jest jasno opisane. I pierwszą rzeczą, o której myślimy, projektując badanie kliniczne, to jest wybór wielkości próby. I oczywiście ta wielkość próby będzie inna dla badania pierwszej fazy, drugiej fazy i trzeciej fazy, bo nie powiedziałam wcześniej, że oczywiście oprócz tego, że trochę na innych rzeczach się skupiamy podczas tych trzech faz do rejestracji, ale też one różnią się populacyjnie. Czyli oczywiście w badaniu pierwszej fazy zależnie od tego, w jakiej jednostce chorobowej przeprowadzamy badanie kliniczne, to może być kilkanaście bądź kilkadziesiąt osób, druga faza to będzie kilkaset, a trzecia faza to nawet kilka tysięcy. Tak jak powiedziałam, jest to zależne oczywiście od populacji, jak bardzo…
K.G.: Dana choroba jest powszechna?
E.B.: Dokładnie tak. Więc zaczynamy od wielkości próby. To też ma związek z tym, żeby jak najmniej ograniczyć ekspozycję pacjentów czy uczestników badania klinicznego na daną substancję, której tak naprawdę nie wiemy, a w badaniu pierwszej fazy bardzo mało wiemy, wiemy tylko i wyłącznie, znamy tylko i wyłącznie wyniki badań na zwierzętach. Więc tutaj organizm ludzki jest wyjątkowy, więc różne sytuacje mogą się zdarzyć. Druga rzecz to jest zaplanowanie użycia placebo, jak będziemy porównywać ewentualnie tą nową terapię. Trzecia rzecz to są punkty końcowe, bo to jest bardzo ważne: co będziemy mierzyć, na jakiej podstawie będziemy oceniać, czy ta nowa cząsteczka jest skuteczna, czy jest efektywna. Kolejna rzecz to są kryteria włączenia i wyłączenia, bardzo ważne, dlatego że kryteria włączenia i wyłączenia dbają o bezpieczeństwo pacjenta. Chcemy na tyle te kryteria przewidzieć, ustalić, żeby zapobiec narażaniu pacjentów, tych, którym możemy zrobić dużą krzywdę, włączając ich, zapraszając ich do udziału w badaniu klinicznym.
K.G.: Czyli wyklucza się część pacjentów.
E.B.: Tak, wyklucza się część pacjentów, dla których udział w badaniu klinicznym byłby zbyt ryzykowny.
K.G.: Byłby większym ryzykiem niż po prostu zwykła terapia.
E.B.: Tak, niż korzyść z tego badania. Kolejna rzecz, oczywiście to jest, planuje się sposób monitorowania bezpieczeństwa, czyli w jaki sposób będziemy monitorować bezpieczeństwo, czyli jak będziemy sprawdzać, czy ten lek jest bezpieczny. Na jakich etapach, co będziemy zgłaszać, które rzeczy będziemy, na co będziemy zwracać na przykład szczególną uwagę. Więc tutaj jeżeli chodzi o to monitorowanie bezpieczeństwa, mamy bardzo dużo narzędzi. Cały ten etap w realizacji badania klinicznego jest bardzo ściśle regulowany. I tutaj na to wszyscy, i sponsorzy, i regulatorzy, mówiąc ogólnie, i komisje bioetyczne bardzo zwracają na to uwagę podczas opiniowania badania klinicznego. Kolejne rzeczy to są procedury randomizacji. W jaki sposób będziemy randomizować pacjentów? Jak będziemy stratyfikować te grupy?
K.G.: Randomizować, czyli?
E.B.: Czyli przydzielać do określonej grupy. Jedna grupa powiedzmy…
K.G.: Czyli chodzi o to, żebyśmy nie zbadali danego leku wyłącznie na kobietach w wieku 40 lat, tylko różnych?
E.B.: Jeszcze nie.
K.G.: A, to nie ten moment?
E.B.: Też jeszcze nie. To właśnie to jest ta stratyfikacja. Czyli np. mamy dwie grupy, randomizujemy do grupy, która będzie otrzymywać cząsteczkę, czyli produkt badany, a druga będzie otrzymać albo placebo, albo najlepszą terapię wspomagającą, albo standardową terapię. To też zależy od badania. Tak że tutaj mamy. Natomiast to, o czym pani powiedziała, to jest właśnie ta stratyfikacja, czyli my będziemy dbać o to, żeby w każdej z tych grup mniej więcej znalazła się odpowiednia liczba kobiet, osób starszych, powiedzmy, osób, które mają choroby współistniejące. Tak że o to chodzi, żeby po prostu nie było tak, że w jednej grupie są tylko kobiety albo jest zdecydowana przewaga kobiet. Oczywiście mamy badania chociażby w ginekologii, gdzie trudno włączyć pana, ale tam, gdzie możemy, to staramy się, żeby ta grupa była między sobą jak najbardziej homogenne, natomiast wewnątrz jak najbardziej heterogenne. Nie wiem, czy to…
K.G.: Jeszcze raz, tak. Między sobą…
E.B.: Między sobą homogenne, czyli żeby one między sobą były podobne. Czyli że mamy podobną, powiedzmy, w pierwszej grupie mamy podobną liczbę kobiet, podobną liczbę mężczyzn, podobną liczbę osób, które mają choroby współistniejące.
K.G.: Czyli w pierwszej grupie tej, która dostanie testowaną cząsteczkę?
E.B.: Tak. I w drugiej, która nie dostanie, żeby było podobnie, prawda?
K.G.: Mniej więcej lustrzane, można sobie to tak wyobrazić.
E.B.: Dokładnie. Żeby można było porównać, żeby te wnioski, które będziemy wyciągać, były jak najbardziej porównywalne. Natomiast w ramach tych grup staramy się, żeby to były grupy heterogenne, czyli żeby właśnie było i kobiety, i mężczyźni, i osoby, które mają choroby współistniejące. To oczywiście zależy od protokołu, ale tak to wygląda.
K.G.: A jest jeszcze coś takiego jak próba podwójnie zaślepiona, to znaczy, że i pacjenci nie wiedzą, czy przyjmują placebo lub już stosowaną terapię, i lekarze też tego nie wiedzą, żeby się nie narzucały wnioski. To się stosuje?
E.B.: Tak, to się stosuje i to się stosuje bardzo często, w zasadzie większość badań trzeciej fazy, czyli tych badań przed rejestracją, to są właśnie podwójnie ślepa próba, tak to się nazywa, podwójnie ślepa próba, czyli ani pacjent, ani lekarz nie wie, czy dany uczestnik otrzymuje produkt badany, czy standardową terapię, czy placebo. Tak jak pani powiedziała, chodzi o to, żeby ograniczać stronniczość.
K.G.: Czy zasugerowanie się, bo to jest potężny psychiczny mechanizm.
E.B.: Tak, zresztą ten efekt placebo w medycynie też znamy, że podawanie czasami placebo i przekazanie uczestnikowi czy pacjentowi, że to jest lek…
K.G.: Patrz: homeopatia, no nie?
E.B.: Prawda? Dokładnie. Zresztą przy lekach przeciwbólowych u osób uzależnionych czasami tak właśnie ten efekt placebo jest bardzo dobrze widoczny, że mówi się, że podajemy lek, podajemy placebo i pacjenta przestaje boleć. Także ten efekt placebo jest udowodniony.
K.G.: Cały odcinek o efekcie placebo, też właśnie w kontekście przede wszystkim leczenia bólu, z profesorem Przemysławem Bąblem w Radiu Naukowym.
E.B.: O!
K.G.: Kojarzy pani, znana postać w tym temacie.
E.B.: Tak.
K.G.: Jeszcze powiedzmy to, że badania kliniczne, wydaje mi się, że one się bardziej kojarzą z takim poszukiwaniem zupełnie nowych leków na nieuleczalne choroby. Ale to pewnie jest jakiś odsetek. Rozumiem, że cały czas szuka się np. lepszych leków na cukrzycę, lepszych leków na nadciśnienie.
E.B.: Tak. Lepszy lek ma szerokie znaczenie, bo to może być lek, który działa tak samo dobrze, ale na przykład ma mniej skutków ubocznych. Albo tak samo dobrze, ma mniej skutków ubocznych, ale jest na przykład rzadziej podawany, bo sposób podawania też nie jest bez znaczenia, zwłaszcza jeżeli chodzi o leki doustne.
K.G.: Tabletka co tydzień, a nie zastrzyki trzy razy dziennie.
E.B.: Na przykład. Albo tabletka raz dziennie, albo tabletka raz na tydzień niż na przykład codziennie. Bo tutaj jeżeli chodzi o leczenie doustne, to należy pamiętać, że im to leczenie pacjent otrzymuje dłużej, ten compliance, czyli przestrzeganie zaleceń, jest coraz słabsze. I to często właśnie tak wygląda, więc tworzenie nowych postaci leków czy leków, które możemy rzadziej przyjmować, jest naprawdę ważne.
K.G.: Mówiła pani o tych wszystkich sprawach, które trzeba opisać na etapie projektowania, czyli jak będziemy sprawdzać bezpieczeństwo, kiedy, jak będziemy oceniać skuteczność. No to jestem sobie firmą farmaceutyczną, projektuję takie badanie, no i dobra, powiedzmy, że zadbam o bezpieczeństwo, ale dość nisko zawieszę sobie poprzeczkę, gdzie uznam, że już lek jest skuteczny. Czy jest ktoś, kto powie: nie no, droga firmo, to jest w ogóle, trochę przeginacie. Tak się nie robi. Nie wydamy zgody na przeprowadzenie takiego badania. Tak jest? Musi być zgoda?
E.B.: Musi być.
K.G.: Czy może być tak, że firma ma na tyle dużo pieniędzy, że przyjdzie do jakiegoś szpitala i powie, słuchajcie, Mercedes tutaj się znajdzie dla kogoś, a tutaj robimy badanie kliniczne. Czy można to zrobić poza systemem?
E.B.: Nie można, nie można. Właśnie chciałam powiedzieć, oczywiście nie można. Dlatego, że mamy odpowiednie regulacje, które bardzo jasno mówią, jaki musi być proces. Czyli również proces realizacji badania, ale proces uzyskiwania zgody na przeprowadzenie badania klinicznego. Oczywiście mamy instytucje międzynarodowe, mamy instytucje europejskie. Każdy kraj członkowski, jeżeli mówimy o Unii Europejskiej, ma swój urząd, w Polsce też, Urząd Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych, który odpowiada za wydanie pozwolenia na realizację badania klinicznego. I oprócz urzędu jeszcze pozytywną opinię dotyczącą badania klinicznego wydaje niezależna komisja bioetyczna. Tak że mamy dwa organy, które czuwają nad tym, żeby, tak jak pani powiedziała, sponsor nie omijał pewnych wytycznych, nie przychodził do badacza czy do ośrodka i mówił, mam tutaj badanie, proszę o zrealizowanie go. Proszę pamiętać o tym, że w Polsce jeżeli badacz będzie prowadził badanie bez zgody Urzędu Rejestracji, ponosi za to odpowiedzialność karną.
K.G.: To nielegalne.
E.B.: Tak, jest nielegalne i po prostu może skończyć w więzieniu.
K.G.: I to też warto podkreślić: pacjenci nie płacą za udział w badaniach klinicznych. To jest czerwona flaga, jeśli kiedykolwiek państwo o tym usłyszą.
E.B.: Tak, to należy uczulać pacjentów. Badania kliniczne, które są prowadzone zgodnie z wytycznymi GCP, zgodnie z Good Clinical Practice, czyli dobra praktyka kliniczna, po angielsku i po polsku, slangowo, i z regulacjami polskimi i międzynarodowymi, za takie badanie po prostu pacjent nic nie płaci.
K.G.: A ta niezależna komisja bioetyczna, bo to dobrze brzmi.
E.B.: Tak.
K.G.: Ale co to znaczy? Kto to jest? Od kogo oni są niezależni? Kto płaci osobom, które pracują w takiej komisji?
E.B.: Oczywiście to jest różnie zorganizowane w różnych krajach, ale w Polsce wygląda to następująco. Po pierwsze, kto to jest? Ona jest niezależna od nikogo, ani od sponsora, ani od badacza, ani od ośrodka. To jest pierwsza rzecz. Czyli niezależna od osób, które mogłyby mieć interes w tym, żeby takie badanie przeprowadzić.
K.G.: Czyli nie może być na przykład zatrudniona w takiej firmie.
E.B.: Dokładnie tak. Właśnie tutaj mamy konflikt interesów, czyli taka osoba, jeżeli…
K.G.: A jeśli żona albo mąż jest zatrudniony na przykład?
E.B.: To też tak.
K.G.: Też to jest wykluczone, tak?
E.B.: Tak, to jest wykluczone. W tej chwili mamy ustawę o badaniach klinicznych produktów leczniczych. Ta ustawa z 2023 roku powołuje Naczelną Komisję Bioetyczną. Oczywiście i w ustawie mamy jasno wpisane, kto może być członkiem Naczelnej Komisji Bioetycznej i w ogóle komisji bioetycznej. I też jest jasno uregulowany konflikt interesów, czyli tam jest jasno wpisane, jaki mamy konflikt interesów. I oczywiście członkami Naczelnej Komisji Bioetycznej i komisji bioetycznych są też lekarze, którzy też są badaczami, bywają badaczami. I wtedy jeżeli coś takiego mamy, to taki lekarz czy lekarka nie może brać udziału w głosowaniu, nie może brać udziału w wydawaniu opinii. bo wtedy oczywiście tutaj mamy ewidentny konflikt interesów. Jeżeli faktycznie żona, która też może być lekarką, czy mąż odwrotnie, prowadzi jakieś badanie i ta osoba, która jest członkiem komisji bioetycznej widzi, że to jest badanie, w którym żona bierze udział, to znaczy jako oczywiście badacz bądź współbadacz, to musi to zgłosić. Oczywiście ta odpowiedzialność leży po stronie członków komisji bioetycznych. Niemniej to jest weryfikowane. I oczywiście tak to wygląda, czyli niezależni od interesariuszy, czyli od sponsora, badacza i ośrodka, czyli od osób, które miałyby w tym interes, żeby to badanie prowadzić.
K.G.: Czyli w Polsce jest jedna taka komisja bioetyczna, która opiniuje te projektowane badania?
E.B.: W Polsce jeżeli chodzi o badania kliniczne produktów leczniczych, mamy Naczelną Komisję Bioetyczną i głównie ona wydaje opinie dotyczące realizacji badań klinicznych. Natomiast ustawa też pozwala na to, żeby były tak zwane komisje uprawnione, czyli komisje, które działają przy instytutach, przy uniwersytetach, przy izbach lekarskich. Jeżeli spełniają określone wymogi, zostaną wpisane na listę komisji uprawnionych. to wtedy też mogą opiniować badania kliniczne produktów leczniczych.
K.G.: A to są duże ciała? Bo im większe, tym trudniej na kogoś wpłynąć, nie?
E.B.: Tak, są różne. Ustawa jasno reguluje liczbę członków. W tej chwili jeżeli chodzi o Naczelną Komisję, to jest 30, mamy tam, także to jest zespół, który składa się oczywiście z dużej liczby osób. Po drugie, są tam przedstawiciele nauk prawnych, przedstawiciele oczywiście nauk medycznych, przedstawiciele nauk etycznych i przedstawiciele pacjentów. I to jest to novum, które zostało wprowadzone.
K.G.: Tego nie było kiedyś.
E.B.: Tak, tego nie było, więc w Naczelnej Komisji Bioetycznej mamy trzech członków, którzy są przedstawicielami pacjentów. Myślę, że to w ogóle zmieniło podejście do opiniowania, w myśl zasady „nic o nas bez nas”, prawda? Czyli pacjenci widzą, jak wygląda zgoda chociażby. Oni bardzo często na to zwracają uwagę. Czy zgoda jest napisana językiem zrozumiałym, prawda?
K.G.: Zgoda, czyli dokument, który widzą pacjenci.
E.B.: Tak, czyli informacja tak naprawdę, zgoda trochę skrótowo, czyli informacja dla pacjenta o badaniu klinicznym i oczywiście później zgoda. Czy to jest czytelne? Bo profesjonalistom często…
K.G.: Czy rozumiemy, na co się zgadzamy.
E.B.: Tak, bo profesjonalistom często pewne rzeczy są oczywiste. Co więcej, my teraz rozmawiamy również z pacjentami, które już mają kilkuletnie doświadczenie, i oni czasami też mówią, że po prostu wpadamy w pewną rutynę, prawda? Jeżeli wpadamy w rutynę, to czasami trudno nam jest zauważyć pewne niuanse. Tak że tak to wygląda, ale to jest na pewno bardzo dobre posunięcie, żeby do zespołów opiniujących włączyć i obowiązkowo musi być przedstawiciel pacjentów.
K.G.: A naczelna komisja jest przy Urzędzie Rejestracji?
E.B.: Przy Agencji Badań Medycznych.
K.G.: Przy Agencji Badań Medycznych. Czyli Agencja Badań Medycznych, proszę wybaczyć, że tak wprost zapytam, ale ta Agencja Badań Medycznych, publiczna instytucja, płaci tym osobom za pracę?
E.B.: Nie, za to płacą sponsorzy.
K.G.: A, to już brzmi gorzej dla ludzi.
E.B.: Tak. Dlatego że sponsor wnosi opłatę do Urzędu Rejestracji, do Naczelnej Komisji Bioetycznej, za opiniowanie tego wniosku.
K.G.: Czy jest różnica, jeśli opinia będzie negatywna a pozytywna?
E.B.: Nie, nie, nie. Sponsor od razu tak jakby uiszcza tę opłatę. To znaczy w momencie, kiedy składa wniosek w takim systemie europejskim, bo do tego jest przygotowany elektroniczny system, Clinical Trial Information System, gdzie sponsor składa całą dokumentację dotyczącą badania klinicznego i wtedy uiszcza opłatę, nie wiedząc jeszcze, jaki będzie finał tego wniosku. Oczywiście to opiniowanie jest procesem i w trakcie tego procesu i komisja etyczna, i Urząd Rejestracji mogą zadać szereg pytań sponsorowi. Sponsor ma określony czas na odpowiedź na te pytania i jeżeli te odpowiedzi nie są wyczerpujące i wystarczające dla osób, które opiniują, wtedy oczywiście takie badanie uzyskuje negatywną opinię i jest odmowa.
K.G.: I to się zdarza?
E.B.: Tak.
K.G.: A mniej więcej jak często?
E.B.: Nieczęsto, dlatego że jakość tych wniosków, które trafia do oceny Komisji Bioetycznej i do Urzędu Rejestracji jest coraz lepsza. Wszyscy się uczymy, więc sponsorzy również. Jeżeli chcemy, żeby lek został wprowadzony na rynek, a taki jest cel, jeżeli sponsor włożył w to wysiłek i pieniądze, nie ukrywajmy, w realizację badań przedklinicznych, to po prostu zależy mu też na tym, żeby badanie było tak zaprojektowane, żeby udowodnić skuteczność i bezpieczeństwo tego leku, prawda? Więc tutaj byłoby nielogicznym, powiedziałabym, przygotowywanie jakościowo złych dokumentów.
K.G.: Załóżmy, że projekt był w porządku i przeszedł pozytywną opinię przez komisję i rozumiem, że zaczyna się właśnie pierwsza faza badań. I wracam do tego, jak rekrutowani są pacjenci do tego badania. Jeszcze raz podkreślamy, nie płacą za to, ale skąd się w ogóle biorą?
E.B.: Biorą się z placówek medycznych. Później, już jak oczywiście badanie uzyska pozytywną opinię komisji, zgodę Urzędu Rejestracji, oczywiście za badanie odpowiada badacz.
K.G.: Badacz, czyli firma czy lekarz?
E.B.: Lekarz. To lekarz deklaruje, jaką liczbę pacjentów jest w stanie włączyć do danego badania. Pracuje w szpitalu, wie, że na przykład miesięcznie, prawda, widzi 30 pacjentów chorujących na cukrzycę powiedzmy, i kryteria włączenia spełnia powiedzmy 20. Wtedy deklaruje, że w przeciągu miesiąca jestem w stanie do badania włączyć 20 czy 10. Oczywiście to zależy. I to są właśnie pacjenci, których lekarz włącza do badania, ale…
K.G.: Po uzyskaniu ich zgody.
E.B.: Oczywiście, po poinformowaniu po pierwsze o ryzykach, o korzyściach, jakie pacjent może odnieść, uczestnicząc w tym badaniu. Natomiast włączanie pacjentów i zapraszanie pacjentów do udziału w badaniu klinicznym odbywa się na podstawie kryteriów włączenia i wyłączenia, czyli oprócz tego, że po pierwsze, lekarz zaproponuje pacjentowi udział w badaniu klinicznym. Po drugie, pacjent zapozna się z informacją, dostanie wyczerpujące odpowiedzi, porozmawia z rodziną, może skonsultować to z innym lekarzem, czyli ma pewność i podejmuje w pełni świadomą zgodę na to, że chciałby wziąć udział w tym badaniu, to oczywiście mamy jeszcze kryteria włączenia i cały proces screeningu. Czyli to, że my zgodzimy się na udział w badaniu, podpiszemy zgodę, nie znaczy, że zawsze do tego badania wejdziemy, dlatego że możemy nie spełnić któregoś z kryteriów włączenia do badania. I mamy tak zwany etap screeningu, czyli etap badań przesiewowych. Bo możemy na przykład, pacjent może nie wiedzieć, że na przykład, nie wiem, ma podwyższoną bilirubinę, która wyklucza go z udziału, dlatego że podanie danej cząsteczki, czyli tego produktu badanego, może jeszcze zwiększać poziom bilirubiny w organizmie i byłoby to dla niego bardzo toksyczne. Tak że tak to wygląda. Czyli generalnie głównie źródłem włączania pacjentów, jeżeli tak możemy powiedzieć trochę karkołomnie, do badań klinicznych są oczywiście placówki medyczne i główny badacz, czyli lekarz.
K.G.: A to jest tak, że takie badanie kliniczne przeprowadza się na przykład w jednym szpitalu czy w różnych?
E.B.: Różnie, różnie. Czasami, jeżeli mówimy tutaj o badaniach klinicznych firm farmaceutycznych, to są najczęściej badania międzynarodowe, wieloośrodkowe, czyli po prostu w wielu krajach i w wielu ośrodkach. Czyli w Polsce może być na przykład 6, 7, 3, to oczywiście zależy od tego, w jakim wskazaniu, czyli w jakiej chorobie, prowadzimy takie badanie kliniczne.
K.G.: Czyli do polskiej komisji, cofając się troszkę, przychodzą nie tylko polskie firmy farmaceutyczne, ale też te międzynarodowe.
E.B.: Międzynarodowe, duże, globalne firmy farmaceutyczne, tak.
K.G.: Przechodzimy do drugiej fazy. Co się dzieje?
E.B.: W drugiej fazie sprawdzamy skuteczność, czyli mamy potwierdzone bezpieczeństwo. W drugiej fazie sprawdzamy skuteczność i ewentualnie dawkę, jeżeli ona jeszcze nie została ustalona w pierwszej fazie. I głównie chodzi właśnie o sprawdzenie skuteczności. Czyli mamy pewne punkty końcowe, gdzie na podstawie tego będziemy sprawdzać, że lek jest skuteczny. I na przykład w onkologii może to być odpowiedź na leczenie, czyli na przykład brak progresji. Czyli w momencie, kiedy mamy zmianę nowotworową, guz, podajemy to leczenie i oczywiście obserwujemy, wykonujemy badanie obrazowe, badanie tomografii komputerowej na przykład. I wtedy sprawdzamy, że ten guz, wymiary tego guza się zmniejszają. Wtedy na podstawie tego możemy uznać, że dany produkt badany ma szansę na to, żeby stać się lekiem, bo jest skuteczny.
K.G.: Tutaj pytanie od jednej z Patronek Radia Naukowego, bo patroni, patronki wcześniej dostają informacje o planowanych nagraniach i u nas na grupie na Facebooku (tajnej!) mogą zadawać pytania. I pani Malwina pyta, czym się różni skuteczność od efektywności?
E.B.: W kontekście badań klinicznych skuteczność to oczywiście jest potwierdzona skuteczność w badaniu klinicznym. Należy też podkreślić jedną rzecz, że w badaniu klinicznym mamy do czynienia z dosyć taką wyselekcjonowaną grupą uczestników. Natomiast w momencie kiedy lek jest rejestrowany i zaczynamy stosować lek w dużej populacji, czyli po prostu u wszystkich pacjentów, którym możemy ten lek podać zgodnie ze wskazaniami, to tutaj mówiłabym właśnie o efektywności. I wtedy…
K.G.: To dość wysublimowana różnica, muszę przyznać.
E.B.: Tak, jest trochę wysublimowana ta różnica. Czyli skuteczność w badaniu klinicznym, efektywność w całej populacji. Ja bym to tak najbardziej może opisowo wyjaśniła w ten sposób.
K.G.: Powiedzmy, że druga faza przebiegła pomyślnie w tym sensie, że okazało się, że badana cząsteczka jest skuteczna lub bardziej skuteczna niż wcześniej stosowane terapie, i przechodzimy do tej trzeciej fazy.
E.B.: Tak, to tutaj właśnie w tej trzeciej fazie po pierwsze porównujemy do standardu, czyli mamy jakiś standard leczenia. Czyli to o czym mówiliśmy, że mamy jakiś lek na cukrzycę, który wiemy, jak działa. W fazie trzeciej będziemy porównywać do tego standardu bądź do placebo, jeżeli nie mamy żadnej opcji terapeutycznej w danym wskazaniu. Po pierwsze na większej grupie osób, bo należy pamiętać, że druga faza to też jest mniejsza liczba uczestników i nie wszystkie na przykład działania niepożądane leku, jeszcze wracając do bezpieczeństwa, nie wszystkie, bo oczywiście druga faza jest głównie skupiona na skuteczności, ale też również badamy, tak jakby bezpieczeństwo jest badane we wszystkich fazach. I teraz tak: nie wszystkie działania niepożądane leku mogą nam się ujawnić podczas tej drugiej fazy, bo mamy zdecydowanie niższą liczbę uczestników niż w badaniu trzeciej fazy. Więc badamy bezpieczeństwo w zdecydowanie większej populacji i porównujemy z tak zwanym standardem leczenia, czyli z tą terapią, którą dostalibyśmy, idąc normalnie do lekarza w ramach, powiedzmy, ubezpieczenia w Polsce.
K.G.: W momencie kiedy, tak sobie wyobrażam, oby nie, ale zachorowałabym na jakiś rzadki nowotwór, który nie idzie leczenie, no to w te pędy będę biegła na badania kliniczne, będę chciała się do nich dostać. Zresztą jeszcze o to będę panią pytać, czy jest jakaś baza właśnie badań klinicznych, do których można by chcieć się dostać. Ale ciekawi mnie, jak weryfikuje się szczepionki. To znaczy w momencie kiedy, no tak jak z tym nowotworem, no to się bada, że powiedzmy guz nie urósł. No to mamy informację. A jak zweryfikować szczepionkę? Czy dane osoby się wystawia na działanie czynnika chorobotwórczego, żeby zobaczyć, w jaki sposób przejdą daną chorobę po szczepionce? Jak to się robi?
E.B.: Chodzi o to, żeby zbadać immunogenność w jakimś sensie, prawda? Czyli podajemy albo żywe, albo w jakiś sposób uszkodzonego wirusa, i wtedy sprawdzamy, jak organizm na to reaguje, czyli nie wystawiamy, no bo samo podanie szczepionki jest w jakiś sposób wystawieniem, prawda?
K.G.: Tak, to rozumiem. No i tutaj mamy ten etap bezpieczeństwa i powiedzmy, że udaje się, że szczepionka jest bezpieczna, jest w porządku, ale jak sprawdzić, czy ona jest skuteczna? To mnie ciekawi.
E.B.: Jak by to powiedzieć? Miernikiem jest…
K.G.: Poziom przeciwciał?
E.B.: Tak, który powinien chronić nas przed tym, żeby nie zachorować.
K.G.: Czyli to nie jest tak, że potem część tych osób się wstawia do pokoju i psika się w nich wirusami czy bakteriami, i potem zobaczymy, jak sobie poradzicie?
E.B.: Nie, nie, nie. Właśnie chodzi o to. Wytworzenie przeciwciał, czyli po prostu liczba przeciwciał, która powinna nas chronić przed tym, żeby zachorować. Czyli w momencie, kiedy organizm jest na tyle odporny, czyli ta liczba przeciwciał powoduje, że nie zachorujemy.
K.G.: No i ta faza czwarta, wracając, kiedy już jesteśmy po rejestracji leku, na czym polega właśnie weryfikowanie w dużo większych grupach, jak to wyłapać w ogóle, że na przykład coś poszło nie tak, że jednak jest jakiś skutek uboczny, na jedną na 10 tysięcy osób.
E.B.: Na przykład. Tak, to prawda. Producent leków, firma farmaceutyczna, bo to są producenci leków najczęściej, jest zobligowany monitorować bezpieczeństwo. To znaczy, my jako pacjenci, użytkownicy, czyli osoby, które przyjmują dane leki, możemy zgłosić tak zwane działania niepożądane. Czyli jeżeli ja przyjmuję lek jakiś, chociażby przeciwbólowy, który przeczytaliśmy, przeczytałam ulotkę, czyli mówiąc fachowo charakterystykę produktu leczniczego, czyli tą ulotkę dla pacjenta, gdzie jest opisane, co się może wydarzyć, a co nie powinno. I oczywiście widzę, że u mnie występują jakieś zdarzenia niepożądane, które nie są opisane w tej ulotce, to wtedy mogę to zgłosić do sponsora, mogę to zgłosić do Urzędu Rejestracji w Polsce. Tak że lekarze tak samo, no bo są leki, które się podaje tylko w warunkach szpitalnych, więc oczywiście pacjent nie zawsze wie, jakie są działania niepożądane tego leku, jakie są skutki uboczne. Więc oczywiście i lekarze, personel medyczny, i pacjenci, wszyscy możemy zgłaszać informacje dotyczące działań niepożądanych skutków ubocznych. I wtedy na tej podstawie sponsor czy Urząd Rejestracji może zdecydować o tym, że po prostu informuje ogólnie społeczeństwo o tym, że ten lek jest toksyczny, i wtedy taki lek jest wycofywany. Albo że powoduje skutki uboczne, albo jest aktualizowana ta ulotka dla pacjenta, także żeby po prostu poinformować o wszystkich działaniach niepożądanych danego leku już wtedy w fazie czwartej.
K.G.: No i tu znowu zapytam, czy to się zdarzało, bo zaczęłyśmy od tej historii z talidomidem, to były lata 50.?
E.B.: 50., 60.
K.G.: A później? Dochodziło do takich sytuacji, gdzie na przykład lek przeszedł już te wszystkie fazy i dopiero w tej czwartej się okazało, że on jednak w masowej populacji może powodować jakieś niebezpieczne rzeczy?
E.B.: Tak, był taki lek przeciwbólowy o nazwie Vioxx, który zdecydowanie zwiększał ryzyko wystąpienia zdarzeń sercowo-naczyniowych. I właśnie ten lek wycofano. To był początek, wydaje mi się, to były lata 2000, może 2005? Na pewno początek lat 2000. I ten lek po prostu został wycofany. I po wycofaniu tego leku też zdecydowanie bardziej zaostrzono monitorowanie działań niepożądanych produktów leczniczych, badanych właśnie w ramach badań klinicznych.
K.G.: To naprawdę są pojedyncze przypadki?
E.B.: Akurat tutaj o tym pamiętam, bo to były początki, kiedy ja zaczynałam zajmować się badaniami klinicznymi, ale lekiem, który też był wycofany, był taki lek na otyłość na przykład, Meridia chyba się nazywał, który też w badaniach porejestracyjnych odkryto toksyczność bodajże wątrobową, jeżeli dobrze pamiętam, ale tutaj nic sobie nie dam uciąć, to musielibyśmy to zweryfikować. Natomiast ta toksyczność była nieakceptowalna i został wycofany.
K.G.: Ale chodzi mi o to, w skali tego, bo my tego nie wiemy. Ile rocznie na polski rynek jest rejestrowanych nowych produktów leczniczych?
E.B.: Rocznie prowadzi się około 700-800, bo to zależy, spływa wniosków o rozpoczęcie badania klinicznego. Chyba w zeszłym roku było rekordowo ponad 800, albo dwa lata temu, wniosków o rozpoczęcie badania klinicznego. Więc to jeżeli chodzi o badania kliniczne, to jest taka skala, natomiast ostatecznie tych cząsteczek może być kilkadziesiąt rejestrowanych, niekoniecznie refundowanych, bo to są dwie różne rzeczy, prawda, rejestracja i refundacja, żeby też tego nie mylić. Bo oczywiście lek może być zarejestrowany, ale niekoniecznie musi być refundowany w Polsce.
K.G.: Czy widzi pani, tak szczerze, jakieś punkty, w których ten system ma jeszcze słabości? Bo proszę wybaczyć, że ja zadaję takie dużo pytań pełnych nieufności, ale one są w społeczeństwie bardzo powszechne. Być może zrozumiałe. Jesteśmy tą firmą farmaceutyczną. Wyłożyliśmy kupę pieniędzy na to, żeby wytworzyć nową cząsteczkę. Wszystko wskazuje na to, że ona zadziała, ale na przykład właśnie już na tych badaniach klinicznych, powiedzmy na drugiej fazie, okazuje się, że ten lek nie jest tak skuteczny, jak byśmy chcieli. I może się pojawić pokusa, żeby trochę te wyniki podkręcić. I zastanawiam się, czy na przykład na tym etapie ten system jest szczelny, czy to jeszcze jest jakoś potem weryfikowane. Jak to działa?
E.B.: Tak, tutaj chodzi, żeby zobiektywizować te wyniki i żeby sponsor nie mógł wpływać na rezultat, to mamy te narzędzia, które tak jakby w samym projektowaniu badania, czyli randomizacja, badania zaślepione. To są tak naprawdę wytyczne, które są przygotowane przez urzędy regulacyjne, żeby właśnie unikać tego wpływu sponsora na badanie, czyli po prostu są wprowadzone oczywiście mechanizmy, można je ulepszać. Kolejną rzeczą, która jest zupełnie niezależna od Urzędu Rejestracji i od komisji bioetycznej, to są właśnie tak zwane niezależne komitety monitorujące, komitety naukowe, gdzie to nie są członkowie, którzy są związani ze sponsorem, prawda? Czyli to jest zupełnie grupa niezależnych ekspertów, która mówi, że drogi sponsorze, twoje wnioski są takie, ale według nas, według naszej opinii, te wnioski są zbyt optymistyczne i nie należy dalej prowadzić, rozwijać tej cząsteczki, dlatego że nie pozwalają na to wyniki drugiej fazy, prawda? I przeprowadzanie trzeciej fazy jest zupełnie nieistotne. Ale też to, że sponsorzy mają obowiązek publikować protokoły badań klinicznych, Czyli jeżeli wejdziemy sobie do tej europejskiej bazy Clinical Trial Information System, czyli Europejska Baza Badań Klinicznych, to tam znajdziemy kryteria włączenia, wyłączenia, znajdziemy protokoły. Oczywiście nikt tam nie ujawnia informacji poufnych, dotyczących chociażby jakichś takich danych handlowych, no bo to też nie o to chodzi. Zresztą w badaniach klinicznych takich danych też nie ma. Można po prostu zobaczyć, jak ten protokół jest zaprojektowany. Kolejna rzecz to też są audyty, inspekcje. Audyt oczywiście przeprowadza sponsor. Może wynająć zewnętrzną firmę, ale to też jest na zlecenie sponsora, więc tutaj to ryzyko jest duże, ale sponsor też musi sprawdzić, czy to badanie jest prowadzone zgodnie z protokołem i zgodnie ze standardami realizacji badań klinicznych. Natomiast inspekcja już jest przeprowadzana przez urzędy państwowe. W Polsce to jest Urząd Rejestracji Produktów Leczniczych. I wtedy to też pokazuje, że to badanie, nawet już później po wydaniu zgody na realizację tego badania, to badanie musi być prowadzone w określony sposób. Jeżeli zdarzają się tam błędy, to można takie badanie po prostu zamknąć.
K.G.: Ciekawa jestem, jak działa technicznie ta podwójnie zaślepiona próba, to znaczy przychodzą te leki gdzieś tam zapakowane, bez oznaczeń? Jak to zrobić, żeby tutaj nie było jakiegoś takiego napisane ołówkiem, słuchaj, to jednak jest ten nasz lek, to tutaj zanotuj, że pacjent A jednak na to skutecznie odpowiada. No ja przepraszam też tutaj lekarzy, że takie są sugestie, ale dużo tu jest hajsu do ugrania.
E.B.: Oczywiście, że tak. Oczywiście, ja rozumiem. Więc tak, jeżeli chodzi o podwójnie ślepą próbę, to najczęściej jest tak, że mamy pacjenta, który podpisał zgodę na udział w badaniu klinicznym. I mamy system elektroniczny, gdzie wpisujemy określone kryteria i takiego pacjenta randomizujemy. System pokazuje nam, czy ten pacjent został przydzielony do grupy A, czy B. I teraz nie wiemy…
K.G.: Czy lekarz to wie?
E.B.: Nie. To znaczy nie wiemy, czy w grupie A jest lek badany, czy produkt badany, czy placebo.
K.G.: Okej, nie wiemy. W ten sposób to jest schowane.
E.B.: Jeżeli przychodzi produkt badany do apteki szpitalnej, powiedzmy, że mówimy o badaniu, które jest prowadzone w warunkach szpitalnych, to przychodzi pacjent na wizytę i teraz również w systemie elektronicznym wprowadzamy numer, bo każdy pacjent w badaniu ma swój unikatowy numer. Wprowadzamy ten numer i system pokazuje nam: proszę wydać pacjentowi trzy opakowania leków o numerach. Operujemy tak naprawdę tylko i wyłącznie numerami. Co więcej, wszystkie te produkty badane i placebo, jeżeli mówimy o badaniu z placebo, są identyczne.
K.G.: Wizualnie.
E.B.: Tak, wizualnie. Tak że ani osoba, która pracuje w aptece nie wie, czy pod danym numerem, w danej fiolce czy w blistrze, mamy placebo, czy produkt badany.
K.G.: A co z badaniami klinicznymi, które są finansowane nie przez firmy farmaceutyczne, a przez instytucję publiczną na przykład? Czy ABM prowadzi, Agencja Badań Medycznych, prowadzi właśnie swoje badania kliniczne?
E.B.: Badania klinicznego jeszcze nie mamy. Mamy eksperyment jako Agencja Badań Medycznych.
K.G.: A czym to się różni?
E.B.: Jeżeli mówimy o badaniu klinicznym, to każde badanie jest eksperymentem, natomiast nie każdy eksperyment jest badaniem klinicznym. Badanie kliniczne to jest pewna interwencja. Jeżeli mówimy o badaniu klinicznym produktów leczniczych, to oczywiście jest podanie pewnej substancji. Natomiast eksperyment nie wiąże się z tak zwaną interwencją, jeżeli chodzi o badanie substancji. I akurat w Agencji Badań Medycznych w tej chwili prowadzimy eksperyment dotyczący testu na wykrywanie endometriozy. Pobieramy fragment tkanki i sprawdzamy po prostu, czy mamy do czynienia z endometriozą, czy nie. Natomiast Agencja Badań Medycznych finansuje badania kliniczne, niekomercyjne, bo te badania, o których pani tutaj wspomniała, to są tak zwane badania kliniczne niekomercyjne, czyli badania, których celem nie jest uzyskanie korzyści, celem nie jest wprowadzenie danego produktu do obrotu, a zwiększenie tak naprawdę wiedzy medycznej. Te badania niekomercyjne są prowadzone przez instytucje, są to uniwersytety, instytuty medyczne czy generalnie placówki, które po prostu zajmują się leczeniem, zajmują się nauką. I te niekomercyjne badania kliniczne najczęściej prowadzone są w tych obszarach, którymi nie interesują się, mówiąc krótko, firmy farmaceutyczne. Dlatego że, powiedzmy, prowadzenie tych badań nie jest zwyczajnie opłacalne. Bo jeżeli mamy małą populację, to wtedy firma farmaceutyczna może skalkulować, że po prostu wydanie pieniędzy na przeprowadzanie badań klinicznych nie jest ekonomiczne, bo po prostu później ta korzyść finansowa jest niewspółmierna.
K.G.: Dlatego leki na choroby rzadkie są tak koszmarnie często drogie?
E.B.: Są tak koszmarnie drogie, bo nawet jeżeli wiemy, że wyprodukowanie tego leku, ten proces wcale nie jest drogi, to oczywiście…
K.G.: Czyli nie jest droższy od takiego innego, nie wiem, na nadciśnienie na przykład lepszego, tak?
E.B.: Tak.
K.G.: Tylko mamy dużo ludzi z nadciśnieniem.
E.B.: Tak, tak, dokładnie, więc po prostu ten aspekt ekonomiczny tutaj jest istotny. I po prostu nawet tak jak mówię, jeżeli sam proces wyprodukowania nie jest drogi takiego leku dla choroby rzadkiej, to po prostu trzeba, mówiąc uczciwie, sfinansować ten cały proces, o którym mówiłam wcześniej, czyli od laboratorium do rejestracji. To jest proces bardzo długi i bardzo kosztowny.
K.G.: No to może tu jest właśnie miejsce na gracza publicznego, który intensywnie będzie współfinansował na przykład takie badanie cząsteczek na rzadsze choroby.
E.B.: Tak, jeżeli mówimy o badaniach niekomercyjnych, to głównie to są wskazania właśnie w tych chorobach rzadkich. I Agencja Badań Medycznych jest taką instytucją rządową, która właśnie finansuje niekomercyjne badania kliniczne. I wypełniamy tę lukę, która była niezagospodarowana do tej pory, bo agencja funkcjonuje krótko ponad 5 lat. Do momentu powstania agencji w Polsce tych badań było dosłownie kilka rocznie rejestrowanych, niekomercyjnych badań klinicznych. W tej chwili to jest kilkadziesiąt, no ale jeszcze daleko nam do Europy Zachodniej, bo w Polsce to jest około, powiedzmy, 10-12%, w Europie Zachodniej to jest powyżej 20%, w Skandynawii nawet 40%, więc mamy co robić.
K.G.: Bo Agencja Badań Medycznych tak brzmi, jakby była od zawsze, ale to nie jest bardzo stara instytucja.
E.B.: Tak, pięcioletnia powiedzmy. Tak, tak, tak.
K.G.: Czy w czasie oceny leku bierze się również pod uwagę np. samopoczucie pacjentów? Myślę tutaj szczególnie o onkologii, dlatego że np. dany lek, wyobrażam sobie, może być bardzo skuteczny w hamowaniu nowotworu, ale z drugiej strony może powodować właśnie non stop, nie wiem, wymioty, biegunkę, trwające tygodniami, że ta jakość życia, no życie trwa, ale jakość jego jest np. nie do zniesienia. Czy to się bierze też pod uwagę?
E.B.: Tak, bierze się pod uwagę, bo oczywiście mówimy o punktach końcowych, czyli to, co będziemy uważać i na podstawie czego będziemy uważać, że lek jest skuteczny. Czyli na przykład lek jest skuteczny dlatego, że nie nastąpiła progresja choroby, jeżeli mówimy o chorobie nowotworowej. Lek jest skuteczny dlatego, że wydłużył całkowity czas przeżycia. Coraz częściej punktem końcowym może też być jakość życia i jest jakość życia. Czyli lek jest skuteczny, ale również jest na tyle bezpieczny, że nie obniża jakości życia. I to jest ważne w chorobach nowotworowych. Kiedyś było tak, że tak naprawdę tą jakość życia oceniało się najczęściej w badaniach trzeciej fazy, czyli tych badaniach przedrejestracyjnych. Natomiast ta jakość życia zaczyna się oceniać teraz w badaniach już drugiej fazy, tam gdzie mówimy o skuteczności, od razu staramy się sprawdzać, na ile ten lek będzie obciążający w takim stosowaniu. Badanie fazy pierwszej jest tak naprawdę zwolnione z oceny jakości życia, natomiast w większości ta jakość życia jest po prostu sprawdzana i monitorowana.
K.G.: Czy jeśli jesteśmy pacjentami bądź bliskimi osób, które właśnie cierpią na jakąś chorobę, czy to rzadką, czy na przykład nie idzie leczenie onkologiczne, czy jest tak, że mamy liczyć na naszego lekarza, lekarkę, że jest zorientowany w temacie, że dzieją się takie badania, czy jest jakieś takie miejsce, infolinia, gdzie możemy zadzwonić i powiedzieć: my bardzo chętnie zapiszemy się na takie badanie kliniczne, bo być może uda się w ten sposób czy wyleczyć, czy zahamować chorobę. No właśnie, czy jest takie miejsce, gdzie możemy jako pacjenci aktywnie szukać takich badań?
E.B.: Chyba najlepszym źródłem wiedzy o badaniu klinicznym powinien być lekarz prowadzący, ale bywa z tym różnie, trzeba powiedzieć. Natomiast mamy tak zwane wyszukiwarki badań klinicznych, czyli miejsca, gdzie możemy sprawdzić, czy na przykład mamy dostępne badania kliniczne w chorobie, na którą cierpimy. Takimi wyszukiwarkami są, największą to jest clinicaltrials.gov. Te wyszukiwarki, akurat to jest wyszukiwarka największa, można powiedzieć, że jest amerykańska, i tam znajdziemy wszystkie badania kliniczne, które są realizowane na świecie. Wadą tego jest to, że ono jest w języku angielskim, więc to jest dużo ograniczenia, no bo język jest ograniczeniem, jeżeli go nie znamy.
K.G.: Tym bardziej taki, medyczny.
E.B.: Tak, tym bardziej medyczny, który sam w sobie może być niezrozumiały nawet po polsku, to prawda. Kolejną wyszukiwarką to jest wyszukiwarka europejska i to jest właśnie wyszukiwarka, która jest w języku polskim. Zresztą ta wyszukiwarka i ten system jest uaktualniany i w ostatnich tygodniach on został uaktualniony. Mamy już tę wyszukiwarkę we wszystkich językach krajów członkowskich, również po polsku. Ja wczoraj wchodziłam w tę wyszukiwarkę, bo chciałam zobaczyć, jak ona funkcjonuje i na ile faktycznie jest przyjazna pacjentom, i w porównaniu do tego, co było jeszcze kilka miesięcy temu, faktycznie ona jest dużo lepsza niż była. Możemy sprawdzić informacje, wpisując na przykład frazę „cukrzyca”, „rak płuca”. I wtedy pokazują nam się ośrodki w Polsce, które realizują te badania. Mamy nazwę badania, mamy ośrodek, gdzie on się znajduje, czyli mamy adres, nazwę i adres ośrodka. Mamy imię i nazwisko głównego badacza oraz mamy numer telefonu, gdzie możemy dowiedzieć się, czy to badanie nadal rekrutuje. Bo też należy pamiętać o tym, że te informacje, nie da się tak tych informacji aktualizować w czasie rzeczywistym. Może być taka sytuacja, że w systemie będziemy mieli informację, że to badanie jest jeszcze w trakcie rekrutacji, to znaczy, tak jak pani tutaj mówi, pacjenci myślą, że można się do niego zapisać. Zadzwonimy i może się okazać, że właśnie wczoraj ta rekrutacja się zamknęła, więc należy dzwonić i o to dopytać. Należy też pamiętać o tym, że to, że my znajdziemy dla siebie, powiedzmy, badania kliniczne, które wydaje nam się, że spełniamy kryteria, żeby zostać wyłączonym do takiego badania, to może się okazać, że jednak do tego badania się nie dostaniemy, dlatego że to o czym mówiłam: te kryteria włączenia, nie spełnimy na jakimś tam etapie tych kryteriów włączenia. Także o tym też należy pamiętać. Mamy też polskie wyszukiwarki. Mamy, ABM też ma wyszukiwarkę swoich projektów, których finansuje, czyli to jest ta wyszukiwarka niekomercyjnych badań klinicznych. Także też można tam ewentualnie szukać badań klinicznych dla siebie.
K.G.: Linki oczywiście do tych wyszukiwarek zostawimy wam w opisie podcastu, gdziekolwiek nas słuchacie, na którejkolwiek z platform. Natomiast tak sobie myślę po ludzku, że np. odmowa zapisania się na takie badanie w przypadku, kiedy mamy do czynienia z jakąś chorobą szybko postępującą, śmiertelną, a na przykład być może testowany jest przełomowy lek, wydaje się być trochę nieludzkie.
E.B.: Tak, ale należy pamiętać o tym, że badanie kliniczne to nie jest coś, co się wszystkim obligatoryjnie należy.
K.G.: Ale to mówi pani z perspektywy technicznej.
E.B.: Tak. To ja powiem, z czego to wynika.
K.G.: Ale tak po ludzku, to myślę sobie, jakbym zadzwoniła w środę, bo we wtorek nie dałam rady, i się okazuje, że się nie dostanę, a tam gdzieś testują coś, co może mi uratować życie, no to trochę serce pęka, nie?
E.B.: Serce pęka, ale też to, co już też zostało poruszone, że w różnych ośrodkach to badanie może rekrutować, czyli w innych ośrodkach. Zawsze można dopytać o to i tam, jak pacjent wejdzie, czy ktoś z bliskich, który też może szukać oczywiście dla bliskiej osoby badania klinicznego, pokazują się ośrodki. To nie jest zawsze tak, że we wszystkich ośrodkach to badanie jest zamykane, rekrutacja jest zamykana jednoczasowo. Możemy szukać innych badań, więc to też czasami faktycznie jest tak, że mamy tylko i wyłącznie jedno badanie w danej jednostce chorobowej. Natomiast należy pamiętać, że tutaj nie mamy na to wpływu po prostu. Badanie jest zamknięte i tyle. Niestety to brzmi bardzo brutalnie, tak jak pani mówi, ale takie są zasady. Należy też pamiętać o tych kryteriach włączenia, że wcześniej Urząd Rejestracji, komisja bioetyczna decyduje o tym, że korzyści przewyższają ryzyka, a w już takiej bezpośredniej sytuacji lekarz–pacjent to lekarz decyduje o tym, czy włączyć danego pacjenta do badania, czy nie. Dlatego że my nie mamy tutaj potwierdzonego bezpieczeństwa i należy o tym pamiętać, nie mamy potwierdzonego bezpieczeństwa danego produktu badanego. Może być tak, że włączenie danego pacjenta do badania może mu przynieść więcej szkody niż korzyści i to trzeba mieć z tyłu głowy.
K.G.: A kwestia placebo. Ja wiem, że to są raczej sytuacje rzadkie, no bo wiele z tych nowych leków jest testowanych w przypadku chorób, na które już jest jakaś terapia. Ale wracam do tej najbardziej dramatycznej sytuacji, która myślę, że też wielu naszym słuchaczom może się gdzieś tam w głowach wyświetlać. No i jest cząsteczka, być może skuteczna. Udało się zapisać na to badanie i co? I dostanę placebo? I ja będę umierać, a ktoś przeżyje?
E.B.: Tak, brzmi to dramatycznie. Badania z placebo stosujemy tylko i wyłącznie wtedy, kiedy nie mamy, tak jak pani powiedziała, żadnej terapii. I może tak być, że nie mamy żadnej terapii. Pojawia się możliwość uczestniczenia w badaniu klinicznym i badanie zawiera placebo, no bo ono ze względów etycznych nie bardzo może zawierać coś innego, bo tak naprawdę to nie jest potwierdzone bezpieczeństwo. I musimy porównać, musimy mieć dwie grupy, żeby zobaczyć: dobrze, jednym podamy, innym nie podamy. Ale może też się okazać tak, że tym, którym podamy, ono wręcz zaszkodziło, więc chronimy tę grupę z placebo. Ale jeżeli się okazuje w trakcie badania, w trakcie badania mamy też coś takiego, możemy w jakimś etapie badania ocenić skuteczność, w trakcie jeszcze trwania badania. Jeżeli się okazuje, że niezależny komitet, ten, o którym na przykład mówiliśmy już, prawda, ocenia, że podawanie tego produktu badanego zdecydowanie poprawia, że choroba się cofa, zmniejsza, zatrzymała, prawda, bo są różne sytuacje. To wtedy może zdecydować o tym, że badanie jest odślepione i wszyscy pacjenci, wszyscy uczestnicy tego badania otrzymują produkt badany, bo niepodanie tej cząsteczki tym pacjentom jest po prostu nieetyczne. Są też takie badania, gdzie podajemy produkt badany i placebo i oceniamy, że… Nie wiemy tego, badanie jest zaślepione i nie wiemy, który pacjent dostaje produkt, a który pacjent dostaje placebo. Wykonujemy tomografię komputerową, oceniamy, że nastąpiła progresja choroby, możemy pacjenta odślepić i wtedy, jeżeli pacjent otrzymywał placebo, włączyć go do grupy, która otrzymuje produkt badany, tak żeby dać mu szansę na to, żeby on jednak otrzymał produkt badany. W momencie, kiedy obserwujemy progresję, odślepiamy pacjenta, uczestnika, okazuje się, że otrzymywał produkt badany, no to okazało się, że on jest nieskuteczny i wtedy takiego uczestnika wyłączamy. Więc to są takie sytuacje, kiedy podajemy placebo. To są rzadkie sytuacje. Placebo też podajemy w sytuacjach, kiedy np. w onkologii, w leczeniu uzupełniającym. Co to znaczy? To znaczy, że w momencie, kiedy mamy standardowe postępowanie, czyli mamy powiedzmy nowotwór piersi i standardem postępowania w takim stopniu zaawansowania jest tylko i wyłącznie operacja, leczenie chirurgiczne. Pacjentka przechodzi operację i przechodzi do obserwacji, czyli monitorujemy, robimy USG czy robimy mammografię, czy tomografię, w każdym razie standardowe monitorowanie stanu pacjentki. Ale wiemy, że oczywiście nowotwory mają tendencję do nawracania i chcemy zapobiegać temu. I możemy włączyć tzw. leczenie uzupełniające. I wtedy te badania też są z placebo, bo nie wiemy, czy np. mamy jakiś lek, który został zarejestrowany, nie wiem, w trzecim stopniu zaawansowania, czyli wtedy, kiedy mamy przerzuty, powiedzmy, czy w czwartym. Ale wiem, że on jest skuteczny na te przerzuty, mówiąc najbardziej obrazowo, ale jeżeli byśmy chcieli udowodnić, że on również zapobiega nawrotom choroby, to też musimy zrobić grupę kontrolną, czyli części pacjentek podać ten produkt badany, którego skuteczność wykazaliśmy w czwartym stopniu, i porównać do grupy, której nie podajemy, bo może się okazać, że po pierwsze on w ogóle nie ma wpływu na nawroty, czyli nie powoduje, że tych nawrotów jest mniej, a jednocześnie może obniżać jakość życia.
K.G.: Bardzo to wszystko jest złożone, bo łatwo się mówi na tabelkach, faza, to, tamto, owamto, numery pacjentów, a kiedy myślimy sobie o panu Robercie, pani Kasi, mamie, tacie, siostrze, to to zupełnie inna dyskusja, czy pacjentowi z perspektywy lekarza.
E.B.: Tak, my też jako agencja staramy się, żeby te badania kliniczne po pierwsze były przyjazne dla pacjentów. Przyjazne to jest takie szerokie pojęcie. Oczywiście nie mamy wpływu na pewne ramy, standardy i pewną formę badania klinicznego, bo ono po prostu musi mieć, żebyśmy się też wszyscy czuli bezpieczni i żeby po prostu ograniczyć jednak, mówiąc tak bardzo brzydko, ekspozycję pacjentów, uczestników wtedy, kiedy ona nie jest konieczna. Staramy się, żeby ten język, którym mówimy do pacjentów, był jak najbardziej czytelny. Pacjenci, grupy, organizacje pacjenckie, grupy pacjentów zaangażowane w przygotowanie protokołów, tak żeby z tych protokołów wyrzucić to, co nie musi się wydarzyć, tak żeby protokół przygotować i zoptymalizować, mówiąc najogólniej. Czyli rozmawiamy z pacjentami, którzy cierpią na daną jednostkę chorobową, żeby zobaczyć, czy z ich punktu widzenia on jest wykonalny. Tutaj jest szereg rzeczy, my oczywiście moglibyśmy bardzo długo mówić, ja mogłabym przytaczać różne przykłady, ale to znaczenie pacjentów i ich głos, ich pewna sprawczość jest coraz bardziej zauważana, coraz bardziej wykorzystywana i pożądana po prostu, żeby angażować pacjentów w takie sytuacje dotyczące projektowania i realizacji badań klinicznych.
K.G.: Tutaj chodzi o jakieś organizacje pacjenckie?
E.B.: Tak, chodzi o organizacje pacjenckie.
K.G.: A to może działać też na tym etapie, że organizacja pacjencka przychodzi np. do ABM-u czy jakiejś innej siostrzanej instytucji w innych krajach i mówi: słuchajcie, my mamy np. chorobę przewlekłą i w tej chorobie najbardziej przeszkadza nam to i to. Czy możecie jakoś zakomunikować w jakiejś firmie farmaceutycznej? Czy mamy problem taki, że nie wiem, mamy nadwrażliwość na coś tam, nie? I spróbujcie nam to jakoś ogarnąć. W tym sensie, że nie że firma się zastanawia nad tym, jaki zrobić lek, tylko pacjenci mówią: potrzebujemy na to, pomóżcie.
E.B.: Tak, tak. I są takie sytuacje, my mamy nawet w tych konkursach, które są organizowane przez Agencję Badań Medycznych, my zachęcamy naszych beneficjentów, czyli badaczy, szpitale, do tego, żeby angażowały organizacje pacjenckie jako konsorcjanta. Czyli wtedy oczywiście oni rozmawiają z organizacją pacjencką. Organizacja pacjencka jest zaangażowana w proces, ma do wykonania określone zadania, chociażby informować o danym badaniu klinicznym, rozmawiać z pacjentami, wiadomo.
K.G.: Oczywiście mają bazę pacjentów.
E.B.: Mają bazę pacjentów, mogą zwiększyć rekrutację, zwłaszcza w chorobach rzadkich, po to, żeby to badanie szybciej zakończyć, szybciej wyciągnąć wnioski. Jeżeli te wnioski pozwalają na to, żeby wyniki tego badania zmieniły rzeczywistość, to wtedy możemy to wykorzystać. Tak że tutaj ta współpraca z organizacjami i ta wiedza i to doświadczenie pacjentów, organizacji pacjenckich jest naprawdę nie do przecenienia na każdym etapie.
K.G.: Znów Patronka Malwina i myślę, że wiele osób się podpisze pod tym pytaniem. Dlaczego jest tak mało leków sprawdzonych czy też dostępnych dla kobiet w ciąży oraz dzieci? I gdzieś jest ta granica do szóstego roku życia. Przed szóstym rokiem życia to prawie nic nie można, a po szóstym nagle wiele więcej jest możliwości. Z czego to się bierze?
E.B.: To się bierze dlatego, że dzieci są pod szczególną ochroną. Oczywiście kobiety w ciąży tak samo, prawda? Więc jeżeli prowadzimy badanie w populacji kobiet ciężarnych, to wtedy musimy zadbać i o mamę, i o dziecko. I to po prostu warunkuje, że tych badań jest mało, bo rekrutacji też do tych badań jest mało.
K.G.: To znaczy, że ryzyko jest zbyt duże po prostu?
E.B.: Właśnie ryzyko jest zbyt duże po prostu. Mamy do czynienia, tak jak powiedziałam, z dwoma organizmami i z organizmem bardzo młodym, który jest na etapie kształtowania. Więc tutaj po prostu to, że ten wpływ na płód jest bardzo, bardzo może być duży. My nie jesteśmy w stanie przewidzieć skutków, jaki dany produkt badany może mieć wpływ na płód. To jest jedna rzecz.
K.G.: Czyli takie testy robi się wyłącznie w sytuacjach, jakby rozumiem, najwyższej konieczności?
E.B.: Tak. Tak, tak. Tylko i wyłącznie, że na przykład możemy badać tylko wtedy, kiedy nie mamy innego wyjścia. Natomiast żeby tak jakby też sprawdzić, jak dany produkt badany będzie wpływał na ciążę, no to oczywiście możemy robić takie badania na ciężarnych zwierzętach. I wtedy możemy sprawdzać, w jaki sposób dany produkt badany będzie wpływał na płód. Tak że tak to się przeprowadza, natomiast jeżeli chodzi o dzieci, to oczywiście należy pamiętać, że po pierwsze musimy chronić. Korzyści ewentualne, te potencjalne korzyści, bo zawsze należy pamiętać, że mówimy o potencjalnych korzyściach, muszą być zdecydowanie większe niż ryzyko. Oczywiście trudnością tutaj też jest to, co pani powiedziała, że od szóstego roku życia. Oczywiście jest to, że ten organizm jest na etapie rozwoju bardzo szybkiego i organizm jest bardzo dynamiczny. On się rozwija, więc ta zmienność różnych czynników w danym organizmie jest bardzo duża. I z tego to też wynika. I to są właśnie te trudności prowadzenia badań w populacjach pediatrycznych i kobiet w ciąży. Chociaż coraz większą wagę przykłada się do inkluzywności w badaniach klinicznych, czyli o włączaniu, o tym, żeby te badania były różnorodne, o włączaniu tak zwanych grup szczególnych, czyli dzieci, czyli kobiet w ciąży, czyli osób, które mają upośledzoną funkcję wątroby czy nerek. Im mamy bardziej różnorodne grupy w badaniu klinicznym, tym później szansa na to, że w tej fazie czwartej, jeżeli będziemy robić takie badanie porejestracyjne i w takim normalnym użyciu już po rejestracji, wtedy mamy większą szansę, że ten produkt będzie bezpieczny właśnie na tym etapie. Dlatego że im większa różnorodność grupy, tym jesteśmy w stanie wyłapać więcej i sprawdzić, zmonitorować więcej tych czynników.
K.G.: Ale też to myślenie o tym, żeby badana grupa była różnorodna, to też był wynalazek, prawda? Bo słyszy się o tym na przykład, że wcześniej bardzo wiele leków było testowanych na męskich organizmach, a potem się okazywało, że na przykład u kobiet to wygląda inaczej. Mówię tutaj z głowy, ale kojarzy mi się coś takiego w przypadku leczenia chorób naczyniowo-sercowych. Coś takiego mi się kojarzy, że bardziej to było właśnie testowane na mężczyznach. Po pierwsze, czy to prawda, że tak było wcześniej? I czy ten wynalazek właśnie jest stosunkowo nowy, żeby testować jednak na różnych grupach? To, co teraz pani podkreśla?
E.B.: Tak, wcześniej oczywiście standardem było to, że oczywiście do badań najczęściej byli kwalifikowani mężczyźni, zdrowi, powiedzmy. Tak to wyglądało, bo było najłatwiej po prostu prowadzić badania, dlatego że nie mieliśmy tej zmienności, o której mówiłam wcześniej. Kobiety były wykluczone z dwóch powodów. Po pierwsze z powodu ryzyka ciąży, zwłaszcza wcześniej, kiedy antykoncepcja nie była tak skuteczna i tak dostępna, ale oczywiście również z powodu tego, że jednak zmienność hormonalna u kobiet, która zależy też od cyklu, miała istotny wpływ na metabolizm, na wchłanianie leków, produktów badanych jeszcze na etapie badania klinicznego.
K.G.: Czyli taki mężczyzna był, proszę wybaczyć brutalność określenia, takim łatwiejszym modelem, bardziej kontrolowalnym.
E.B.: Tak. Bardziej kontrolowalnym, bardziej homogennym, czyli po prostu mieliśmy taką grupę, gdzie wyniki były zbliżone.
K.G.: Łatwiejsze do porównania?
E.B.: Łatwiejsze do porównania, dokładnie tak. Natomiast teraz to się zmienia, dlatego że widzimy, że im bardziej ta grupa jest różnorodna, tym później, nawet jeżeli mamy pewną trudność na etapie wyciągania wniosków, to później okazuje się, że mamy mniej działań niepożądanych w takim standardowym stosowaniu już później po rejestracji.
K.G.: Pani Magdalena pyta: czasami badania kliniczne są skrócone, jak to było w przypadku pandemii COVID-19. Kto o tym decyduje i co jest brane pod uwagę przy takiej decyzji?
E.B.: Badanie może być skrócone z wielu powodów. Jeżeli chodzi o te badania, które były skrócone z powodu COVID, no to oczywiście to było globalne zagrożenie. Instytucje regulacyjne, rządy państw w Polsce, to jest ministerstwo, mogło podjąć taką decyzję, że takie badanie zostanie skrócone, bo po oszacowaniu ryzyka można było oszacować, czyli założyć, że skrócenie badania, nawet przy tym, że zakładamy, że nie mamy pełnej informacji dotyczącej bezpieczeństwa, i tak będzie mniejszym ryzykiem, niż w ogóle niepodanie danego produktu, które było w tym badaniu, szerszej populacji. Tak że to głównie od tego zależy. Też skrócenie badania może wynikać właśnie, przy COVID poniekąd to było, z braku alternatyw. Są czasami takie sytuacje, że na przykład badania kliniczne zostaje zamknięte po drugiej fazie, bo jeżeli mówimy o tym, że nie mamy żadnego leczenia w danym wskazaniu, to co pani mówiła, że po prostu nie mamy nic i badanie fazy drugiej pokazuje, że ta skuteczność jest bardzo wysoka, no to wtedy możemy rozpocząć proces. I oczywiście do tego są specjalne ścieżki, o których ja tutaj nie będę mówić, no bo to są ścieżki dla sponsorów, co sponsor musi zrobić, żeby rozpocząć po prostu procedurę wcześniejszej rejestracji. I właśnie to też jest w chorobach rzadkich, tam gdzie nie mamy terapii, to są tak zwane choroby sieroce. Wtedy też możemy takie badanie skrócić. Tak jak powiedziałam, decydują o tym instytucje krajowe odpowiedzialne generalnie za zdrowie.
K.G.: Jeszcze jedno pytanie i myślę bardzo ciekawe, pani Anny. Jak to się dzieje, że leki wprowadzone na rynek, żeby leczyć jedno schorzenie, są później wykorzystywane do leczenia czegoś zupełnie innego? Bo to się zdarza przecież. I jakie badania wtedy muszą przejść, pyta pani Anna. I to też mnie bardzo ciekawi, jak w ogóle się wpada na coś takiego? To musi być chyba jakiś przypadek.
E.B.: Nie, nie, nie. Nie zawsze to jest przypadek. Tak zwane leki off-label. Po pierwsze, bywa tak, że my wiemy na przykład, że obecny dany lek na etapie badania pierwszej fazy był na przykład sprawdzany i weryfikowany w różnych wskazaniach. I z jakiegoś powodu sponsor zdecydował, że będzie go rozwijał tylko i wyłącznie w jednym wskazaniu. Bo te powody mogą być bardzo różne, bardzo prozaiczne czasami. Ale my wiemy, że ta cząsteczka ma również potencjał w innym wskazaniu. I wtedy można spróbować oczywiście stosować takie leki, tak zwane off-label, czyli poza wskazaniem. Dlatego że to, że dana cząsteczka jest rejestrowana w określonych warunkach, dlatego że badanie kliniczne było przeprowadzone w określonych warunkach. I sponsor jasno w informacji dla pacjenta o leku jasno musi wyjaśnić, w jakich warunkach i w jakim wskazaniu i w jakich dawkach ten lek może być stosowany. Ale ten lek może po pierwsze być stosowany właśnie u dzieci, może w mniejszych dawkach, prawda? A u dzieci nie był badany, więc on wtedy stosowany jest off-label, czyli poza wskazaniem. U kobiet w ciąży, bo nie był badany u kobiet w ciąży, więc nie możemy napisać, że ten lek może być stosowany u kobiet w ciąży, ale wiemy, że badania przedkliniczne na ciężarnych, powiedzmy, zwierzętach wykazały, że on nie ma działania negatywnego na płód, prawda? Natomiast sponsor w badaniu trzeciej fazy nie prowadził badania na kobietach ciężarnych, no bo to trudno jest po prostu prowadzić takie badanie na kobietach ciężarnych.
K.G.: Ale kto o tym wie i kto decyduje o takim użyciu poza wskazaniem?
E.B.: Naukowcy i najczęściej to są właśnie lekarze naukowcy i najczęściej to są niekomercyjne badania kliniczne. To o czym mówiliśmy.
K.G.: Okej, czyli bierzemy tę samą cząsteczkę i sprawdzamy, bo tak pani powiedziała, że to można użyć poza wskazaniem, ale to chyba nie jest tak, że można sobie w szpitalu użyć: a, to jest do jednego, to…
E.B.: Nie, użyć nie, nie. Musi być zgoda komisji bioetycznej tak naprawdę.
K.G.: Dobra. Przepraszam, że to tak doprecyzowuję.
E.B.: Tak, oczywiście, bardzo dobrze.
K.G.: Bo zrozumiałam, jakby można był o tak, że: a, ja pamiętam, że przecież to było też do czego innego, to użyję to tutaj u tego pacjenta. Tak nie wolno?
E.B.: Nie, nie, nie. To nie można sobie tak wziąć i użyć. Nie można robić tak zwanej samowolki, przepraszam, że tak powiem obrazowo. To też jest regulowane, czyli używanie leków off-label też jest regulowane.
K.G.: Jeśli chcielibyście przeczytać więcej informacji, zobaczyć różne infografiki, to zajrzyjcie proszę też na stronę ABM, gdzie właśnie prowadzona jest też taka akcja edukacyjna, jeśli chodzi o badania kliniczne. Tak zupełnie na koniec chciałam panią zapytać: przez czas pani aktywności zawodowej to ten świat badań klinicznych, on się rozwinął? Czy mamy raczej taką w pewnym sensie stagnację? To znaczy, że no tak, już jakiś czas temu ustaliliśmy odpowiedni standard i się go trzymamy. Jakby pani na to popatrzyła tak z perspektywy, to co mamy?
E.B.: Ten świat się zmienił. Oczywiście, że się zmienił. Ja badaniami klinicznymi zajmuję się już ponad 20 lat i zmieniły się po pierwsze standardy etyczne. Na pewno tak.
K.G.: Czyli?
E.B.: Czyli uczestnik badania klinicznego jest coraz bardziej bezpieczny. Ta ocena, zachowanie godności, ocena bezpieczeństwa…
K.G.: Jest podmiotem, a nie przedmiotem badania.
E.B: Tak, jest podmiotem badania. Sam fakt, że do zespołów opiniujących w komisjach bioetycznych zapraszani są pacjenci, to też jest duża zmiana. To są ostatnie dwa lata. Też podejście do pacjenta, tak żeby to było tak, że nie tak, że my szukamy pacjenta do badania klinicznego, tylko szukamy badania klinicznego do pacjenta. Cała idea, która też jest poniekąd wynikiem pandemii, badań zdecentralizowanych, czyli żeby to badanie dotarło do pacjenta, a nie pacjent do ośrodka, gdzie musi często przebyć bardzo długą drogę kilkugodzinną, żeby dotrzeć do tego ośrodka. Tak że tutaj są różne mechanizmy, o których też możemy długo gadać, jeżeli chodzi o badania zdecentralizowane. Więc ten cały proces jest dynamiczny. Medycyna spersonalizowana i te badania kliniczne też są coraz bardziej spersonalizowane. Nowe cząsteczki są, które mają coraz mniej toksyczności, czyli po prostu tych skutków ubocznych, mówiąc najprościej. Tych czynników, które zmieniają badania kliniczne jest bardzo dużo. My też chcemy w ogóle budować świadomość w społeczeństwie dotyczącą badań klinicznych. I tak jak pani redaktor tutaj zachęcała do odwiedzenia strony Agencji Badań Medycznych, ja oczywiście też do tego zachęcam, ale również zachęcam do tego, żeby odwiedzić stronę „Pacjent w badaniach klinicznych”. I tam państwo możecie znaleźć informacje, bardzo rzetelne informacje, dotyczące badań klinicznych. Bo oczywiście trudno jest opowiedzieć o różnych aspektach, niuansach, bo tak jak państwo pewnie zdążyliście zauważyć, że tych niuansów dotyczących badań klinicznych jest bardzo dużo. Więc ja zachęcam do odwiedzenia tej strony. Można przeczytać, zaznajomić się z tymi informacjami. My też jesteśmy bardzo otwarci na pytania, na sugestie. Zapraszamy do kontaktu z Agencją Badań Medycznych. Często też pacjenci piszą do nas z prośbą o to, żeby powiedzieć, że właśnie pytają, jak znaleźć badania kliniczne. My zawsze staramy się wtedy pomóc. Jeżeli takie badanie jest prowadzone, wysyłamy mailowo listę badań klinicznych, które są na przykład prowadzone w Polsce, z numerami telefonów. Także zachęcamy do odwiedzenia strony Agencji i zachęcamy do odwiedzenia strony „Pacjent w badaniach klinicznych”. Na koniec jeszcze chciałam podkreślić jedną rzecz: że oczywiście w badaniu klinicznym mamy wielu interesariuszy, mamy sponsora, mamy badacza, zespoły badawcze, ośrodek, mamy Urząd Rejestracji, komisję bioetyczną, ale musimy zawsze pamiętać, podkreślam: zawsze pamiętać, że najważniejszym uczestnikiem badania klinicznego jest właśnie uczestnik, jest pacjent. Jest osoba, która dzięki temu, że wyraziła zgodę na udział w badaniu klinicznym, może odnieść korzyści z udziału w tym badaniu klinicznym, ale nie mamy pewności co do tego. To jest osoba, która często ponosi szereg niedogodności jako uczestnik badania klinicznego. I miejmy naprawdę bardzo duży szacunek dla uczestników badań klinicznych, bo oni, tak jak powiedziałam, mogą mieć korzyść albo nie. Natomiast my jako osoby, które…
K.G.: Zachodzące do tych aptek.
E.B.: Tak, zachodzące do tych aptek, dokładnie. Albo będziemy korzystać z tego, że mamy nowy lek, który został zarejestrowany dzięki właśnie uczestnikom badań klinicznych, dzięki temu pacjentowi, który był uczestnikiem, albo ta cząsteczka nie będzie dalej rozwijana i wszystkie siły naukowców, badaczy skupią się na nowych cząsteczkach. Tak że tak naprawdę beneficjentami badań klinicznych jesteśmy my, czyli użytkownicy leków, mówiąc tak bardzo szeroko. Dzięki właśnie tym uczestnikom badań klinicznych.
K.G.: Tak mówiła pani doktor nauk medycznych Elżbieta Bylina, Agencja Badań Medycznych, dyrektorka Centrum Rozwoju Badań Klinicznych. Dziękuję serdecznie.
E.B.: Bardzo dziękuję.
Udostępnij:
Obiecane w odcinku linki:
Dyrektor Centrum Rozwoju Badań Klinicznych w Agencji Badań Medycznych