

Pracuje na Wydziale Matematyki, Informatyki i Mechaniki Uniwersytetu Warszawskiego. Specjalizuje się w algorytmach biologii obliczeniowej, algorytmach randomizacyjnych oraz obliczeniach odpornych na błędy.

Profesor w Baylor College of Medicine w Houston, Teksas, USA. Jego badania skupiają się na lepszym zrozumieniu mechanizmów molekularnych oraz konsekwencji fenotypowych rearanżacji genomu
To mogła być zwrotnica ewolucyjna – mówi w Radiu Naukowym prof. Paweł Stankiewicz, genetyk z Baylor College of Medicine w USA. Razem z prof. Anną Gambin, bioinformatyczką z Uniwersytetu Warszawskiego zajmują się badaniem redukcji liczby chromosomów z 48 do 46 – kluczowej w historii naszej ewolucji. Redukcja spowodowała powstanie definitywnej bariery reprodukcyjnej między liniami ewolucyjnymi naczelnych (nie możemy mieć płodnego potomstwa z gorylami), mogła się przyczynić również do rozwoju intelektualnego i smuklejszej postawy.
– Nastąpiła w wyniku połączenia się dwóch chromosomów, które wciąż są obecne u naczelnych: chromosomu 2A i 2B, w wyniku tzw. fuzji. Analizując miejsce tej fuzji w genomie człowieka w różnych populacjach stwierdziliśmy, że jest on wszędzie takie samo. Metodami bioinformatycznymi i molekularnymi udowodniliśmy, że to zdarzenie wystąpiło w historii naszej ewolucji tylko raz – tłumaczy prof. Stankiewicz.
Ten jeden samiec
Naukowcy zaproponowali model, w którym źródłem tej zasadniczej zmiany był jeden samiec. Redukcja doprowadziła u niego do 47 chromosomów. Ten następnie miał potomstwo z wieloma samicami, które miały klayscznie 48 chromosomów. W kolejnym pokoleniu dochodziło do poronień lub potomstwa z 48 lub 47 chromosomami. Ze względu na krzyżowanie się kuzynów, w trzecim i kolejnych pokoleniach pojawiały się osobniki z 46 chromosomami. Tutaj zobaczycie schemat:
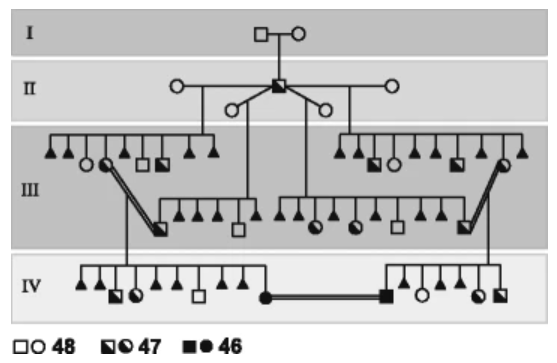
– Błysk w oku, wysportowana sylwetka, mogli zwracać uwagę… – żartuje, ale tylko trochę prof. Gambin, sugerując, że osobniki ze zmniejszoną liczbą chromosomów mogły się ze sobą chętniej krzyżować.
Kiedy to się stało? – Naprawiliśmy błąd matematyczny, który był w istniejącej metodzie i z poprawionych rachunków otrzymaliśmy datę mniej więcej 900 tys. lat temu – wyjaśnia prof. Anna Gambin. Ustalenia prof. Gambin i prof. Stankiewicza były cytowane w niedawnej pracy w Science. Autorzy pracy szacują, że właśnie około 900 tys. lat temu mieliśmy do czynienia z początkiem ewolucyjnego „wąskiego gardła” – na Ziemi miało żyć nieco ponad 1200 osobników naszych przodków, zdolnych do reprodukcji. I to przez 100 tys. lat. Zmiana mogła się dzięki temu utrwalić. – To mógł być tak zwany efekt założyciela – dodaje prof. Stankiewicz
Jednocześnie oboje przyznają, że ich datowanie nie jest (bo nie może być) stuprocentowo pewne. Do fuzji mogło dojść wcześniej. Zagadką pozostaje to, ile chromosomów miał np. Homo erectus, zaliczany do rodzaju Homo, a żyjący już 2 miliony lat temu. Póki co, brakuje jednak odpowiednio zachowanego materiału genetycznego, żeby móc to rozstrzygnąć.
Samą rozmowę zaczynamy od wyjaśnienia zjawiska zmian genomowych – dotyczących całych sekwencji genów, a nie mutacji pojedynczego genu. Takie duże zmiany z jednej strony mogą być przyczyną wielu chorób genomowych, z drugiej prawdopodobnie miały ogromne znaczenie w naszej ewolucji, jako nośnik względnie szybkich zmian.
Hurtowe zmiany w genomie
Pojęcie chorób genomowych zaproponował w latach 90. i przez kolejne dekady rozwijał, prof. James Lupski, amerykański genetyk. Prof. Stankiewicz od lat z nim współpracuje.
W odcinku posłuchacie też o tym, czy ewolucja ludzi dzieje się nadal, dlaczego należy wyrzucić do kosza określenie „śmieciowe DNA”, dlaczego tak kluczowa jest regulacja genów (patrzcie na motyla i gąsienicę – przecież obie formy mają ten sam zestaw genów!), o ile tysięcy lat można się pomylić w modelowaniu przeszłości (i jak to ograniczać), a także dlaczego Amerykanie chętnie współpracują z informatykami z Polski.
Radio Naukowe działa dzięki społeczności wspierającej na https://patronite.pl/radionaukowe Bardzo dziękuję! Mamy kolejne plany rozwoju, jeśli chcesz w nich pomóc – każda kwota ma znaczenie
TRANSKRYPCJA
Karolina Głowacka: W studio Radia Naukowego dwójka gości – pani profesor Anna Gambin. Dzień dobry.
Anna Gambin: Dzień dobry.
K.G.: Pani profesor jest z Wydziału Matematyki, Informatyki i Mechaniki, Instytut Mechaniki Uniwersytetu Warszawskiego. Zajmuje się bioinformatyką, szczególnie w genetyce. Jest z nami także pan profesor Paweł Stankiewicz. Dzień dobry.
Paweł Stankiewicz: Dzień dobry.
K.G.: Który jest genetykiem, pracuje w Baylor College of Medicine w Houston w Stanach Zjednoczonych, konkretnie w Zakładzie Genetyki Molekularnej i Genetyki Człowieka na tejże uczelni. Muszę od razu zapytać, co pan tu robi przy stoliku Radia Naukowego. Nie w Houston, tylko tu, w Warszawie.
P.S.: Mam tutaj rodzinę, mamę, którą odwiedzam przynajmniej parę razy w roku. I przy tej okazji akurat jestem.
K.G.: Zdradzimy od razu, że oboje jesteśmy ze Skarżyska-Kamiennej. Pozdrawiamy wszystkich słuchaczy z tego miasta i z całego świętokrzyskiego. Przed nami prawdziwie interdyscyplinarny odcinek. Temat wielowątkowy – będzie o naszym pochodzeniu jako ludzi, będzie o ewolucji, będzie o genach, genomie, modelach bioinformatycznych. Kawał solidnego odcinka, słuchajcie uważnie. Zaczniemy od genetyki, od wyjątkowości genomu przodków Homo sapiens i tego, co mogło to spowodować w ewolucji. Może na początek, jak ten nasz genom jest skonstruowany? Bo mało osób wie, że niewiele z tego, co jest w naszym DNA, tak naprawdę koduje białka, czyli daje te instrukcje, żeby białko się wytworzyło. Proszę nam o tym opowiedzieć, panie profesorze.
P.S.: Jednym z najważniejszych wniosków z zakończonego ponad dwadzieścia lat temu projektu poznania genomu człowieka było to, że oprócz tych około dwóch procent sekwencji DNA, które kodują białko człowieka, nasz genom składa się w większości z sekwencji powtarzalnych, petytywnych, które w dużej części odpowiadają właśnie za jego niestabilność, za jego kruchość.
K.G.: Ale wtedy to był szok, prawda? Pytano, po co w ogóle ta reszta jest. To słynne śmieciowe DNA.
P.S.: Nie rozumieliśmy wtedy tak dobrze, jak teraz, po co to DNA jest, czy ono jest tylko śmieciowe, czy pełni jakąś funkcję. Ale te powtarzalne sekwencje, które w większości składają się z tzw. retrotranspozonów czy segmentalnych duplikacji, o których powiem później, były również przeszkodą w tym, że to sekwencjonowanie genomu w zasadzie było niedokończone. Dopiero w ubiegłym roku po dwudziestu latach, dzięki zastosowaniu technik sekwencjonowania nowej generacji z długimi odczytami, udało się kompletnie złożyć genom, poznać jego całkowitą strukturę.
K.G.: Czyli dwadzieścia lat później. Na czym polega trudność przyjrzenia się dokładnie temu, jak ten genom jest zbudowany? No bo tutaj właśnie z jednej strony wielki sukces, że mamy sekwencję, a teraz pan nam mówi, że było to jeszcze dokładniej obserwowane.
P.S.: Okazuje się, że te powtarzalne w genomie sekwencje, które w jakimś sensie są dziedzictwem ewolucyjnym, które dostaliśmy w spadku głównie od naczelnych w trakcie ewolucji, są problemem biologicznym dla komórki, ale również technologicznym dla naukowców, ponieważ trudno za pomocą tych dotychczas stosowanych krótkich sekwencji złożyć nasz genom.
K.G.: Co informatyka na to?
A.G.: Można tu podać taki bardzo prosty przykład – może niektórzy państwo lubią układać puzzle. Wyobraźmy sobie przepiękny górski krajobraz do ułożenia z ponad tysiąca elementów. Niestety, trzysta elementów przedstawia błękitne niebo, praktycznie bezchmurne. I to są te nasze powtarzalne elementy w genomie, których nie umiemy dopasować, poskładać w ciąg, ponieważ są praktycznie takie same. Nowa technologia sekwencjonowania polega na dłuższych odczytach. Rozdzielczość jest gorsza – to znaczy, mamy większe elementy puzzli, jak dla małych dzieci. A jeśli na naszym bezchmurnym niebie chociaż gdzieniegdzie jest chmurka albo przelatujący ptak, to takie większe elementy pozwalają zahaczyć o te specyficzne fragmenty i wtedy łatwiej dopasować. Tak samo z długimi odczytami – możemy zahaczyć o pewne charakterystyczne w genomie fragmenty, które już się nie powtarzają, i poskładać.
K.G.: Czyli nie byłoby współczesnej genetyki bez bioinformatyki, nie ma takiej możliwości. Wróćmy w takim razie do tych powtarzalnych elementów, które są w naszym genomie. Po co one są? Dlaczego one się powtarzają? Kojarzyłoby się właśnie, że to śmieciowe, że to jakiś błąd, jakaś pozostałość, zniknie to za jakiś czas.
P.S.: Właśnie w tych ostatnich dwudziestu latach lepiej zrozumieliśmy, że te powtarzalne sekwencje pełnią również ważną funkcję w genomie. I zamiast określać to śmieciowe DNA jako pustynię genomową, jak robiliśmy do tej pory, teraz raczej uważa się, że jest to morze regulacji. I wiemy, że to niekodujące DNA, te powtarzalne sekwencje były prawdopodobnie najważniejszym mechanizmem w ewolucji naszego genomu. Przez tę niestabilność genomu w tej chwili w jakiś sposób uczestniczą one w rozwoju fizjologicznym człowieka i powstawaniu chorób.
K.G.: To powiedzmy o tych chorobach, bo z kolei pana współpracownik, profesor James Lupski, wykazał, że problemy z tymi powtarzalnymi fragmentami naszego DNA mogą być przyczyną konkretnych chorób.
P.S.: W latach dziewięćdziesiątych profesor Lupski w Houston w Teksasie ukuł takie określenie – choroby genomowe. I przez ostatnie ponad trzydzieści lat rozwinął całą tę dziedzinę chorób genomowych. W skrócie chyba najlepiej można to zobrazować, jeśli sobie porównamy nasz genom do książki, która się składa z czterdziestu sześciu rozdziałów, bo mamy czterdzieści sześć chromosomów. W przeciwieństwie do takich klasycznych mutacji punktowych, które polegają na zmianie pojedynczych nukleotydów, pojedynczych cegiełek, w przypadku chorób genomowych dochodzi do utraty lub powielenia, duplikacji całych wyrazów, zdań, paragrafów, a nawet rozdziałów. My to określamy jako zmienność liczby kopii fragmentów DNA, które objawiają się właśnie poprzez delecję lub duplikację. I jeśli w obrębie tych regionów znajdują się tzw. geny wrażliwe na dawkę, może dochodzić do zmian fenotypowych.
K.G.: Czyli tego, jak wyglądamy.
P.S.: Tak. I okazuje się, że prawdopodobnie właśnie te sekwencje powtarzalne, to niekodujące DNA jest bardziej odpowiedzialne za to, co odróżnia nas od naczelnych, od szympansów, niż same sekwencje kodujące.
K.G.: Czy takie choroby genomowe to np. chyba najbardziej znana choroba genetyczna, czyli zespół Downa?
P.S.: W jakimś sensie tak, bo zespół Downa jest powodowany przez dodatkową kopię chromosomu dwudziestego pierwszego.
K.G.: To kawał wyrazów.
P.S.: Tak, cały rozdział jest podwojony. Natomiast najczęściej są to mniejsze zmiany, w zasadzie submikroskopowe – to znaczy, że nie widać ich pod mikroskopem w analizie kariotypu. I często są to zmiany powtarzające się, nawracające. To są takie klasyczne zespoły mikrodelecyjne, mikroduplikacyjne.
K.G.: Czyli albo coś znika, albo się podwaja. Delecja – znikanie, duplikacja – powielanie. To jest szalenie ciekawe, co pan powiedział, że wiele wskazuje na to, że to właśnie ta część naszego genomu odróżnia nas w sposób zasadniczy od naszych krewniaków – szympansów, szympansów bonobo, innych naczelnych. Myślę, że większość naszych słuchaczy kojarzy, że często się mówi, że nasze geny są niemalże takie same, jeśli chodzi o szympansy. W ilu procentach?
P.S.: W dziewięćdziesięciu ośmiu procentach na poziomie białka jesteśmy identyczni.
K.G.: Na poziomie tego kodującego. A cała reszta, to, co miało być tą pustynią, a okazało się, jak pan mówi, morzem regulacji, to jest to, co jest najpewniej najbardziej kluczowe?
P.S.: Wydaje się, że ewolucji łatwiej było manipulować sekwencjami niekodującymi oraz całymi genami, doprowadzając np. do ich duplikacji czy amplifikacji, i eksperymentować raczej na kopii niż na oryginalnej matrycy.
K.G.: W takim razie dobry pomysł na eksperymenty. Czy to w tej części naszego genomu kryje się coś, co jest odpowiedzialne właśnie za tę regulację genów, to, jak one się ujawniają na ekspresję? Pani profesor?
A.G.: Tak. Powiedzieliśmy już, że jesteśmy świadomi, że bardziej niż sama sekwencja genetyczna, to, co wpływa na nasz fenotyp, na zespół cech charakteryzujących dany organizm, to jest regulacja genów, czyli taka dosyć skomplikowana sieć interakcji, która determinuje, które geny, w jakich momentach rozwoju organizmu i w jakich tkankach są tworzone, ulegają ekspresji. Myślę, że dobrym przykładem będą dwa organizmy, które mają identyczną sekwencję genetyczną, np. motyl i gąsienica. Jest to jeden organizm w dwóch wersjach na różnych etapach rozwoju. Ale na pierwszy rzut oka są to zupełnie inne zwierzęta. To, co naprawdę je różni, to jest właśnie regulacja genów. Dodatkowo Paweł wspomniał o transpozonach, o sekwencjach powtarzalnych. One też odgrywają niebagatelną rolę w ewolucji z tego powodu, że występują w bardzo różnych odległych ewolucyjnie od siebie organizmach, w jakiś sposób się przemieszczają. Retrotranspozony pochodzą od retrowirusów, a dodatkowo się namnażają.
K.G.: W tym sensie, że to są całe kawałki DNA, które wplatają się w DNA?
A.G.: Tak, wplatają się w DNA poprzez zakażenie tym wirusem i potem zostają w genomach.
K.G.: My też je mamy, prawda?
A.G.: Oczywiście, że mamy.
K.G.: Jesteśmy hybrydą z wirusami. Profesor trochę kręci głową. [śmiech]
A.G.: Te transpozony również często stanowią miejsca wiązań czynników transkrypcyjnych, czyli takie miejsca w DNA, gdzie przyczepiają się białka uruchamiające cały proces transkrypcji, produkcji innych białek. Więc są szalenie ważne dla tej regulacji.
K.G.: Myślę, że większość osób, które rozmyślają nad ewolucją, nad tym, że zachodzi ona dlatego, że dochodzi do losowych zmian i jedne się okazują dobre, więc zostają w pokoleniach, a inne nie, zastanawia się, jak to się mogło wydarzyć? Oczywiście mieliśmy parę miliardów lat, więc było dużo czasu, ale jednak pojedyncza zmiana miała spowodować zasadniczą różnicę. To jest chyba ten klucz, którego ja się dowiedziałam od państwa wcześniej, że mogą być istotne zmiany właśnie w tych dużych kawałkach. Nie, że zmienia się literka, tylko zmienia się całe zdanie czy cały akapit. Na ile tego typu zmiany w naszym DNA mogły spowodować szybsze zmiany ewolucyjne?
P.S.: Tak jak Ania wspomniała, chodzi o regulację. W przypadku chorób genomowych, kiedy efekt fenotypowy objawia się przez zmianę dawki genu, czyli dawki białka, a nie zmiany jego struktury.
K.G.: Dawki genu? Czyli pojawia się więcej genów i produkuje się więcej białek?
P.S.: Nie. Jeśli mamy dodatkową kopię genu, to oczywiście mamy więcej białka, ale można również zmieniać ekspresję genu poprzez jego regulowanie. Czyli zwiększać produkcję RNA poprzez to białko. To wynika z jego regulacji przez sekwencje regulatorowe, które najczęściej znajdują się właśnie w tym niekodującym DNA, czasami w bardzo dużej odległości.
K.G.: Ten gen robi się np. bardziej aktywny?
P.S.: Tak. A czasami mniej, bo dochodzi do zwiększenia lub zmniejszenia ekspresji. Łatwiej ewoluować, modyfikować, zmieniając tę ilość białka, niż jego strukturę.
K.G.: I pewne cechy są bardzo wrażliwe właśnie na ilości białka?
P.S.: Dokładnie.
K.G.: I one mogły spowodować np. jakieś zmiany skokowe?
P.S.: Czasami są to zmiany skokowe, czasami odbywa się to w wolniejszym tempie. I tak jak skokową zmianą jest powstanie choroby u człowieka, tak samo w ewolucji dochodziło do pewnych zmian, które były jakąś jej zwrotnicą, czyli bardzo dużą zmianą.
K.G.: I tutaj dochodzimy właśnie do tej zwrotnicy, która jest jednym z głównych tematów naszego dzisiejszego spotkania. Mianowicie tego niezwykłego momentu w ewolucji, który najpewniej w pewnym sensie doprowadził naszych przodków do tej ścieżki, którą staliśmy się my teraz. Oczywiście nie taki był plan ewolucji, to nie jest tak, że ewolucja sobie zaplanowała, że stworzy przyszłą podcasterkę, która będzie mówiła przez mikrofon, chociaż byłoby miło sobie tak myśleć. Chodzi o zmniejszenie liczby chromosomów. Panie profesorze, długo myśleliśmy, że mamy tyle samo, co szympansy. Jak to było?
P.S.: Tak. Mało kto wie, że policzenie tego, ile mamy chromosomów, nie było takie proste. Przez ponad trzydzieści lat w pierwszej połowie XX wieku ludzie mylnie uważali, że mamy czterdzieści osiem chromosomów. Dopiero na przełomie 1955 i 1956 roku, czyli trzy lata po odkryciu helikalnej struktury DNA, udało się określić prawidłową liczbę, czyli czterdzieści sześć, a nie czterdzieści osiem. Czterdzieści osiem chromosomów mają małpy człekokształtne – szympans, goryl i orangutan. Wiemy, że w pewnym momencie naszej ewolucji musiało dojść do zmniejszenia liczby tych chromosomów, do redukcji. Zaczęliśmy się zajmować tym tematem kilka lat temu i udało nam się wyjaśnić, kiedy i jak do tego doszło.
K.G.: To może powiedzmy, kiedy, a za chwilę jak do tego doszło. Tutaj zwracam się do pani profesor, bo pani jest od modelowania. To dzięki pani pracy można próbować odpowiadać na takie pytania. Jak spróbowaliście na to odpowiedzieć i co wam wyszło?
A.G.: Użyliśmy pomysłu, który istniał już wcześniej w literaturze. Ludzie zaobserwowali, że są pewne charakterystyczne struktury w DNA, w genomie, na chromosomach, które częściej występują przy ich końcówkach. Mając genom, można policzyć, jaki jest rozkład tych specyficznych mutacji na nim, a następnie popatrzeć, jakby to wyglądało średnio, nie patrząc na końcówki, na środek. I to samo zjawisko występuje zarówno u człowieka, jak i u naszych spokrewnionych małp człekokształtnych. Weźmy chromosom, który uległ fuzji, czyli to miejsce, które się połączyło, występuje teraz blisko środka chromosomu drugiego u człowieka. Powinien pozostać ślad tego, że kiedyś były to końcówki chromosomów. Więc nagromadzenie tych mutacji powinno być wyższe, niż wskazywałoby położenie w środku chromosomu. I właśnie w ten sposób dokonuje się obliczeń. Porównujemy charakterystyczny wzorzec tych mutacji, które obserwujemy, z takim średnim wzorcem, który by nastąpił, gdyby nie było tej fuzji. Im bardziej różnią się od siebie te dwa wzorce, tym mniej czasu upłynęło od fuzji. Bo jeszcze nie zanikł ślad tego, że te chromosomy stanowiły oddzielne byty.
K.G.: Czyli trochę patrzycie, jak świeże jest to sklejenie.
A.G.: Tak. Naprawiliśmy błąd matematyczny, który był w istniejącej metodzie, i z tych poprawionych rachunków otrzymaliśmy datę dziewięćset tysięcy lat temu – wtedy nastąpiła fuzja chromosomu drugiego.
K.G.: Czyli dziewięćset tysięcy lat temu – zapamiętajcie to, drodzy słuchacze, bo wrócimy jeszcze do tej liczby – według obliczeń moich dzisiejszych gości doszło do tej redukcji liczby chromosomów z czterdziestu ośmiu do czterdziestu sześciu. Jak mogło do tego dojść? I czy to się działo wielokrotnie, czy to się wydarzyło raz? Może zacznijmy od tego, jaki był ten mechanizm zejścia do czterdziestu sześciu. Dlaczego? Gdzie tutaj ta pomyłka mogła się pojawić?
P.S.: Wiedzieliśmy, że ta redukcja liczby chromosomów nastąpiła w wyniku połączenia się dwóch chromosomów, które wciąż są obecne u naczelnych, chromosomu 2A i 2B lub dwunastego i trzynastego w wyniku tzw. fuzji, ale wiedzieliśmy, że to nie było takie zupełnie czyste sklejenie, ponieważ nastąpiła również utrata pewnego materiału. Wiemy, przyglądając się kariotypom człowieka i naczelnych, że jednak spory fragment subtelomerowy końcówki chromosomu został utracony. W tych regionach subtelomerowych jest największe stężenie, koncentracja tych segmentalnych duplikacji, o których mówiłem, całych zduplikowanych fragmentów DNA, które stanowią siedem procent DNA człowieka i które destabilizują nasz genom. Analizując miejsce tej fuzji w genomie człowieka w różnych populacjach, stwierdziliśmy, że jest ono wszędzie takie samo. Czyli potwierdziliśmy metodami bioinformatycznymi, ale również molekularnymi przez sekwencjonowanie, że to zdarzenie w naszej ewolucji wystąpiło tylko raz. I to jest troszkę w przeciwieństwie do tzw. translokacji robertsonowskich u człowieka, które są dość podobnym mechanizmem, w wyniku których następuje zmiana liczby chromosomów, czyli tzw. aberracje numeryczne, liczbowe. Są to bardzo częste aberracje u człowieka, co tysięczny człowiek jest nosicielem takiej translokacji robertsonowskiej.
K.G.: W translokacji robertsonowskiej chodzi o to, że te krótkie ramionka chromosomu się sklejają?
P.S.: Spośród dwudziestu trzech par chromosomów człowieka pięć par to są tzw. chromosomy akrocentryczne. To znaczy, mają bardzo krótkie jedno ramię, na którym w zasadzie nie ma żadnych genów. Translokacja robertsonowska to jest właśnie połączenie takich dwóch akrocentrycznych chromosomów w obrębie tych sekwencji krótkiego ramienia lub centromeru.
K.G.: Przypomnijcie sobie, drodzy słuchacze, chromosomy to takie iksy. One mogą być albo symetryczne, albo mieć przesunięcie tego centrum i te ramionka są krótsze. My mówimy o tej sytuacji, kiedy jest niesymetrycznie i są dwa krótsze ramiona chromosomu.
P.S.: Tak. Dobrym przykładem takiej translokacji konsekwencji klinicznych jest też zespół Downa, ponieważ jednym z akrocentrycznych chromosomów jest ten z dwudziestej pierwszej pary. Jeśli taki chromosom zostaje odziedziczony czy powstaje w wyniku takiej translokacji, to mamy jego dodatkową kopię. Te translokacje są powtarzające się i w zasadzie w każdym przypadku mają inne miejsce połączenia, sklejenia. Natomiast w przypadku chromosomu drugiego, który jest produktem tej ewolucyjnej fuzji…
K.G.: I to wiemy na pewno.
P.S.: Tak, jest to wynik naszej analizy. To było zdarzenie jednorazowe, ono się już nie powtórzyło. W wyniku tej zmiany doszło do redukcji liczby chromosomów z czterdziestu ośmiu do czterdziestu sześciu poprzez to wprowadzenie bariery reprodukcyjnej między Homo sapiens a małpami człekokształtnymi, które też mają czterdzieści osiem chromosomów, podobnie jak w przypadku konia, osła i muła, gdzie kombinacja sześćdziesięciu czterech chromosomów i sześćdziesięciu dwóch u osła daje sześćdziesiąt trzy u muła, który jest już niepłodny.
K.G.: Ale skoro miałoby dojść do czegoś takiego raz, to ma pan na myśli, że raz u jednego osobnika, samca czy samicy, jednego z naszych przodków? Po prostu raz jedyny w historii Ziemi?
P.S.: Tak. I najprawdopodobniej było to u samca. To jest oczywiście spekulacja, bo tego nigdy nie wyjaśnimy, natomiast znając podstawy segregacji chromosomów, mejozy, rodowody, i wykorzystując wiedzę z cytogenetyki klinicznej, zaproponowaliśmy model, w jakim się to stało. Klan i rodowód osobników jeszcze nie człowieka, ale przed człowiekiem, w którym prawdopodobnie był jeden samiec, który krzyżował się z wieloma samicami, i doszło do transmisji tej fuzji, która zdarzyła się u niego albo w komórkach rozrodczych, albo była ona może mozaiką, czyli była obecna również w jego komórkach somatycznych.
K.G.: Czyli on już miał czterdzieści sześć chromosomów?
P.S.: Nie. Część jego plemników miało dwadzieścia trzy, a nie dwadzieścia cztery chromosomy, ponieważ dwa się połączyły. I wtedy z połączenia jego dwudziestu trzech chromosomów z dwudziestoma czterema którejś samicy wyszło czterdzieści siedem.
K.G.: To skąd czterdzieści sześć?
P.S.: To był pierwszy etap, bo to było pierwsze pokolenie, to były dzieci tego samca. Natomiast, ponieważ było wiele samic, wiele matek, mogło dalej dochodzić do krzyżowania się dzieci między sobą. Jeżeli sobie wyobrazimy krzyżowanie się osobnika męskiego i żeńskiego z czterdziestoma siedmioma chromosomami, będą częste poronienia, będą w tym rodowodzie powroty do czterdziestu ośmiu chromosomów. Czasami będzie dochodziło do kolejnej redukcji do czterdziestu sześciu chromosomów. I wtedy mamy w drugim pokoleniu po tym samcu dwa osobniki, które mają po czterdzieści sześć chromosomów. Ich krzyżowanie się już wtedy nie jest obarczone żadnymi problemami z niepłodnością i dochodzi do efektu założyciela i rozchodzenia się. To był taki moment w naszej ewolucji określany właśnie pojęciem efektu założyciela. Jak stwierdziliśmy w wyniku naszych badań, prawdopodobnie była z tego jakaś korzyść ewolucyjna. To znaczy, nie tylko ta bariera reprodukcyjna, która raz na zawsze oddzieliła nas od małp człekokształtnych, ale zidentyfikowaliśmy ciekawy gen, czynnik transkrypcyjny FOX T4L, którego dwie kopie uległy utracie w czasie tej fuzji, a ten gen okazuje się w ostatnich latach bardzo ciekawy dlatego, że odgrywa bardzo ważną rolę w rozwoju mózgu u człowieka, ale też u wielu różnych zwierząt. A my stwierdziliśmy jego najwyższą ekspresję w móżdżku i w nerwie piszczelowym, co by mogło pośrednio wskazywać, że utrata kopii czy zmiana ilości białka tego genu mogła być odpowiedzialna za postawę dwunożną.
K.G.: Czyli mogli się stać ad hoc mądrzejsi i chętniejsi do chodzenia w sposób bardziej wyprostowany? Przecież kości itd., jednak nie było tak łatwo.
P.S.: Nie tak łatwo, ale być może był to jakiś czynnik, czyli trochę nam to pomogło. W jakimś sensie swój swojego rozpoznał w tym rodowodzie i ta fuzja miała szansę się rozejść.
A.G.: Bardziej wysportowana sylwetka, błysk w oku i później już byli atrakcyjniejsi. To, co było ważne w tych naszych badaniach, to to, że udało nam się po raz pierwszy poskładać te kawałki chromosomów naczelnych, które w czasie fuzji zostały utracone. Bo wcześniej środowisko naukowe, składając genomy naczelnych, interesowało się odpowiedniością, homologiami, podobnymi rejonami do genomu człowieka. Natomiast najciekawsze dla nas było to, co zostało utracone w czasie fuzji, co, jak pani wspomniała, doprowadziło nas do bycia ludźmi. Było to zupełnie nowe podejście, algorytm, który, wracając do puzzli, nie działa w taki sposób, jak do tej pory robili ludzie, że wszystkie możliwe elementy razem. Wrzucali sobie tysiące tych elementów i próbowali z nich naraz poskładać cały genom. My staramy się zacząć od pojedynczego elementu. Koncentrujemy się np. na rejonie fuzji, to jest nasza kotwica, a później pomalutku rozszerzamy, budujemy w obie strony, wybierając tylko te elementy, które mogą w jakiś sposób do siebie pasować. To jest taki fajny algorytm o dźwięcznej nazwie PhaseDancer autorstwa Basi Poszewieckiej, która właśnie będzie broniła doktorat. Wydaje się, że będzie on miał ogromne zastosowania, nawet w genetyce medycznej, kiedy trzeba lokalnie dowiedzieć się, jak wygląda fragment genomu, który uległ pewnym zmianom z powodu niestabilności.
K.G.: Pani Poszewiecka to pani doktorantka, tak?
A.G.: Tak.
K.G.: Pozdrawiamy serdecznie i trzymamy kciuki. To, o czym państwo mówią, brzmi w sposób naprawdę niezwykle działający na wyobraźnię, że mieliśmy jakiś taki przełomowy moment. Ale ja muszę to dokładnie zrozumieć – dlaczego przekonujecie mnie, że to się wydarzyło tylko raz, skoro, jak pan sam mówił, zdarzają się te zmiany i wiem, że zdarzają się współcześnie osoby, które się rodzą z czterdziestoma pięcioma chromosomami? No to dlaczego nie? Nie do końca rozumiem różnicę między tymi translokacjami robertsonowskimi – przypominam, chodzi o te zmiany, gdzie mamy chromosom, który nie jest symetryczny, tylko te rączki są troszkę krótsze. Nie rozumiem, dlaczego może tak być, że czterdzieści pięć jest teraz współcześnie u kogoś… Jeszcze tych ludzi jest więcej, mogliby się spotkać – dwa razy czterdzieści pięć i dalsza redukcja mogłaby się wydarzać.
P.S.: Chodzi tutaj o mapowanie i sekwencjonowanie tzw. punktu złamania, czyli połączenia dwóch końców chromosomów. Te połączenia powstają czasem w różnych mechanizmach w wyniku błędów replikacji lub rekombinacji. W przypadku translokacji robertsonowskich, o których wiemy, że są powtarzające, nawracające się, ponieważ nie ma ich u rodziców czy dziadków, powstają u nowego osobnika. Są metody, które określają najpierw z mniejszą rozdzielczością te miejsca, te punkty złamań, ale ponieważ one się znajdują w obrębie tych sekwencji powtarzalnych, było bardzo trudno to określić na poziomie jednego nukleotydu. Dopiero w ostatnim roku dzięki zastosowaniu tych technik sekwencjonowania nowej generacji z długimi odczytami udało się zmapować, zsekwencjonować dokładnie punkty złamania. I za każdym razem ten punkt jest inny. Czyli translokacje robertsonowskie wciąż powstają, nasz genom wciąż jest dynamiczny, zmienny. Za każdym razem jest to inna translokacja. Natomiast nasza fuzja chromosomu drugiego, jak analizowaliśmy ten punkt złamania, punkt połączenia dwóch chromosomów i wszystkie warianty w jego okolicy w różnych populacjach, każdy osobnik, każdy człowiek ma dokładnie to samo łącze. Jest on odziedziczony. W związku z tym możemy powiedzieć, że w zasadzie wszyscy obecnie żyjący ludzie musieli pochodzić od tego jednego rodowodu, nie ma innej drogi.
K.G.: To jest grube, tak bym powiedziała. Co na to środowisko? Bo to jest naprawdę mocne twierdzenie. Państwo się uśmiechają.
A.G.: Wydaje mi się, że tej argumentacji chyba nikt nie neguje. Nie da się tego podważyć. Dodatkowo ostatnie, sprzed kilku tygodni wyniki opublikowane w „Science” sugerują, że był taki moment w naszej historii, gdzie na Ziemi przez sto tysięcy lat żyło około tysiąca osobników.
K.G.: Pani profesor znakomicie prowadzi naszą rozmowę. Bardzo za to dziękuję. Dlatego, że faktycznie teraz jest doskonały moment, żebyśmy doszlusowali do kolejnego mocnego akcentu w tym wywodzie. Pamiętacie, mówiłam wam, drodzy słuchacze, żeby zapamiętać dziewięćset tysięcy lat temu, czyli wtedy, kiedy według państwa obliczeń miało dojść do tego połączenia, do tej redukcji genów z czterdziestu ośmiu do czterdziestu sześciu i uruchomienia tej nowej ścieżki ewolucyjnej, której jesteśmy, nie powiem „finałem”, ale etapem. Co się wydarzyło dziewięćset tysięcy lat temu zdaniem badaczy, którzy opublikowali swoje wyniki w „Science”? Pani profesor.
A.G.: Oni nie tyle spekulują, co się wydarzyło. Ich obserwacja to pewien matematyczny model, który umożliwia śledzenie rozwoju populacji ludzkiej, jej dynamiki. Właściwie podzielili naszą historię na bardzo malutkie fragmenty czasu i zastanawiają się, jak liczna była populacja ludzka w tych fragmentach. Sam pomysł na poznanie tej dynamiki jest trochę podobny do tego, jak my szacowaliśmy fuzję. Nasza data fuzji również bardzo pasuje do ich obliczeń, co spowodowało, że im też spodobała się nasza obliczona data. Przede wszystkim starają się zgromadzić dosyć reprezentatywną próbkę genomów żyjących osobników. Rozważyli około trzech i pół tysiąca żyjących teraz ludzi, oglądali ich genomy i koncentrowali się na takich miejscach, gdzie występują z kolei punktowe mutacje, nie takie duże, o których tu rozmawiamy, tylko punktowe typu single nucleotide polymorphism, po polsku snipy. Patrzymy na te snipy i zastanawiamy się, w jakim procencie populacji dany snip jest zmieniony, występuje tam inna literka niż u większości. Wyobraźmy sobie, że mamy w populacji tylko dziesięciu osobników i mamy dziesięć takich miejsc, które obserwujemy. Patrzymy na pierwsze miejsce i patrzymy, u ilu osobników jest zmienione na drugie, na trzecie, na dziesiąte, a następnie całą tę informację koncentrujemy w jeden wektor liczb zliczający, ile jest mutacji u jednego osobnika. Powiedzmy, że trzy. Ile jest mutacji, które wystąpiły u dwóch osobników? Dwie. I tworzymy tzw. widmo częstości mutacji. Mając to widmo, okazuje się, że potrafimy wyprowadzić model matematyczny, który powie nam, jak takie widmo wygląda przy założeniu pewnego modelu ewolucji. To znaczy, przy założeniu, jak kształtowała się liczność naszej populacji. Można to zrobić za pomocą takiej teorii koalescencji. Załóżmy, że czas płynie do tyłu i cząsteczki, czyli genomy łączą się u wspólnego genetycznego przodka. Ten proces można matematycznie opisać i wyprowadzić pewne wzory, jak powinno wyglądać nasze widmo częstości mutacji. Jak mamy ten przypadek z modelu, mamy prawdziwe widmo częstości mutacji, możemy porównać te dwie wielkości i próbować znaleźć parametry modelu, czyli w tym przypadku liczności populacji na poszczególnych etapach rozwoju, żeby jak najlepiej tłumaczyło zaobserwowane wartości, to widmo częstości mutacji, które mamy teraz. Zostało to zrobione. Samo pojęcie widma częstości było już używane w genetyce populacji, natomiast pomysł, żeby podzielić naszą historię na maleńkie fragmenty, żeby to bardzo sprawnie, numerycznie policzyć, jest zasługą autorów tej pracy. I właśnie okazało się, że według nich istniał tak długi przedział czasu, jak sto tysięcy lat, kiedy populacja była ekstremalnie mała – rzędu tysiąca dwustu osobników, praludzi. Zgadza się to również z wykopaliskami, ponieważ brakuje dokładnie z tego okresu szczątków kopalnych praludzi.
K.G.: Nie natrafiliśmy do tej pory. To by się zgadzało, że jeśli było ich tak mało, to mamy małe szanse na to, żeby się zachowali.
A.G.: Tak. Oczywiście samo wytłumaczenie, dlaczego tak się działo, jest bardzo ogólnikowe – susze, zmiany klimatu. Jedna rajska dolina, gdzie ta populacja ma przetrwać. Z drugiej strony, jeśli tam zaszła ta fuzja, to właśnie była szansa, że się utrwaliła.
K.G.: Czyli badaczom z Chin i Stanów wychodzi, że ten bottleneck, czyli wąskie gardło populacyjne, zaczęło się też mniej więcej te dziewięćset tysięcy lat temu. I to się nakłada z waszymi obliczeniami. Wróćmy jeszcze, proszę, na chwilę do tego, jakie mogło mieć to znaczenie. Pan mówił o tym, że jeden konkretny gen jest aktywny szczególnie w móżdżku i w nerwie piszczelowym i on mógł jakoś spowodować większą chęć do dwunożnej postawy. Jakby pan porównał ten moment, co to mogło być? To naprawdę mogło jakoś zasadniczo zmienić, że na pierwszy rzut oka mogli się rozpoznać? Nie wiedzieli, że ten ma czterdzieści sześć chromosomów, a ten czterdzieści osiem, ale że jakoś inne są te osobniki.
P.S.: Można na to patrzeć na trzech różnych poziomach. Rozwój mózgu jako takiego, gdzie utrata tego genu czy zmiana ilości białka z genu FOXD4L mogła powodować większy, szybszy rozwój mózgu u osobników, które miały tę fuzję. Najpierw w tym pierwszym pokoleniu w postaci jednego chromosomu, a potem już dwóch. Bo my mamy genom diploidalny i mamy tak naprawdę dwie fuzje. Czyli efekt jakby się podwoił u osobników, które mają czterdzieści sześć chromosomów. Jeden poziom to jest wpływ na rozwój naszego mózgu. Drugi – stwierdzamy jego wysoką obecność w móżdżku, a móżdżek u człowieka jest odpowiedzialny za koordynację ruchów. Natomiast nerw piszczelowy jest jednym z nerwów, który unerwia mięśnie, które kierują naszą stopą. A więc jeśli połączymy to ze sobą, to przejście z pozycji czterokończynowej na dwunożną wymaga bardzo dużej koordynacji nerwowo-mięśniowej. I to właśnie mogłoby być dzięki zmianom utraty tego genu i lepszej koordynacji móżdżku.
K.G.: I naprawdę tak się widzieli?
A.G.: Nie potrafimy wszystkiego przewidzieć, ale może mogły to być jeszcze jakieś bardziej subtelne sygnały.
K.G.: Że ktoś jest np. bystrzejszy?
A.G.: Tak.
K.G.: To ciekawe. Pan Patryk, jeden z patronów Radia Naukowego – bo dałam wcześniej znać, że będę z państwem rozmawiać – pyta o to, na ile moment wąskiego gardła może powodować, że ten garnitur czy też suknia genetyczna może robić trochę taki chów krewniaczy? Jakie mogą być konsekwencje ewolucyjne tego zawężenia? A pan Grzegorz pyta, czy aby w czasie takiego bottleneck nie jest właśnie łatwiej o powstanie, utrwalenie się mutacji? Trochę o tym mówiliśmy. Pan profesor.
P.S.: W genetyce populacyjnej pojęcie wąskiego gardła jest bardzo dobrze znane, nie tylko w ewolucji genomu człowieka. Następuje najczęściej w wyniku jakiejś katastrofy, kataklizmu ekologicznego, klimatycznego, kiedy dochodzi do znaczącej redukcji liczby osobników. Poprzez to następuje zubożenie puli genowej i najczęściej ma to efekt raczej negatywny, bo jednak dla ewolucji jest korzystniejsze, kiedy ten genom jest bardziej zróżnicowany.
K.G.: Chyba że mamy jakąś bardzo fajną zmianę.
P.S.: Tak, wtedy pojawia się kolejne pojęcie tzw. dryfu genetycznego, kiedy zmiana, która zajdzie w takim momencie, ma szansę rozejścia się później w przyszłych pokoleniach. W skrajnym przypadku, kiedy jest bardzo mała populacja, można mieć do czynienia z tzw. efektem założyciela. Tak jak w tym przypadku być może łatwiej było się potem rozejść, utrwalić w populacji tej fuzji dzięki temu, że startowała z małej populacji, ale również dzięki temu, że miała te korzyści ewolucyjne, o których mówiliśmy, czyli większy rozwój mózgu, móżdżku, postawę dwunożną.
K.G.: Na ile państwo są przywiązani do tej liczby dziewięćset tysięcy lat temu? Czy to jest coś, co jednak mogło się wydarzyć wcześniej, później? Pani profesor.
A.G.: To jest dobre pytanie, bo w momencie, kiedy dokonujemy jakichś obliczeń, w przypadku bioinformatyki kiedy projektujemy procedury obliczeniowe, które coś liczą, zawsze musimy się liczyć z tym, że są jakieś błędy. Informatycy często mówią o numerycznej poprawności i stabilności. Jak wykonujemy bardzo wiele obliczeń, to nawet błędy zaokrągleń mogą się skumulować. Staramy się zawsze oszacować tę niepewność. Tam ta niepewność też jest oszacowana. Niestety przedział ufności i wariancja jest dosyć spora, rzędu kilkuset tysięcy lat.
K.G.: Taki jest margines błędu?
A.G.: Tak.
K.G.: To spory, przyznaję. Pytam dlatego, że wszyscy jesteśmy ludźmi, więc fajnie to brzmi, że wam i badaczom, którzy opublikowali swoje wyniki w „Science”, się to nałożyło, ale porządny naukowiec musi zachowywać dystans emocjonalny i może powiedzieć, że może niekoniecznie. Pan profesor.
P.S.: Jakieś dziesięć lat temu, kiedy udało się zsekwencjonować genomy neandertalczyka i denisowianina dzięki zastosowaniu tych najnowszych technik sekwencjonowania nowej generacji, udało się stwierdzić, że ta fuzja była obecna również w ich genomach.
K.G.: Też mieli czterdzieści sześć?
P.S.: Najprawdopodobniej. A możemy to powiedzieć, ponieważ wiemy, że się z nimi krzyżowaliśmy, było potomstwo, to potomstwo było płodne i w naszych genomach nosimy średnio te półtora procent, w niektórych populacjach jest to nawet pięć, sześć procent genomu denisowian.
K.G.: Denisowian czy neandertalczyków?
P.S.: Neandertalczyków i denisowian. Tylko akurat w przypadku dalekiej Azji tamta pozostałość jest od denisowian. W Europie bardziej neandertalczyków. A ponieważ wiemy, kiedy mniej więcej doszło do rozdzielenia się linii Homo sapiens od neandertalczyka i denisowianina, wiemy, że ta fuzja musiała powstać wcześniej. I to się też fajnie wpisuje w naszą ocenę.
K.G.: A kiedy to było?
P.S.: Około siedmiuset tysięcy lat temu. Wcześniej datowanie tej fuzji różnymi metodami doprowadziło do dużej rozbieżności czasu, właśnie od tych kilkuset tysięcy lat do nawet kilku milionów. I możemy sobie teoretyzować, że jest możliwe, że ta fuzja mogła powstać wcześniej. Mogło to być np. zdarzenie, które nas kompletnie oddzieliło od szympansów.
K.G.: Pytam o to dlatego, że taki Homo erectus zaczął żyć już dwa miliony lat temu. I oczywiście wiem, że to nie jest tak, że od przodka do przodka, że to jest taka drabinka jeden do drugiego, tylko raczej mamy jakieś rozmazane rzeki tych różnych odnóg. Muszę przyznać, że to mnie zaskoczyło. Wiemy, ile miał chromosomów?
P.S.: Nie wiemy i z wielką niecierpliwością na to czekamy. Być może się doczekamy, kiedy naukowcy będą mieli materiał o lepszej jakości DNA, który nie będzie tak zdegradowany. Być może to się dzieje w tej chwili, kiedy tu rozmawiamy. Na pewno by to bardzo pomogło lepiej określić datowanie tej fuzji.
K.G.: Czyli jest możliwe, że to było wcześniej, tak? Nawet dwa miliony lat temu mogło to być możliwe?
A.G.: Jest też możliwe, że Homo erectus nie miał tej fuzji.
P.S.: I w tę, i w tę.
K.G.: Ale dalej będą się państwo upierać, że to się zdarzyło raz. Czy w ewolucji może się pojawić coś takiego po raz kolejny? Że gdzieś u, dajmy na to, samca Homo sapiens dojdzie do takiej redukcji z czterdziestu sześciu do np. czterdziestu czterech? I to się zacznie rozchodzić w populacji.
P.S.: Jak najbardziej tak. Mamy na to przykłady w literaturze naukowej – nieliczne, ale jednak mamy. Przykłady, kiedy identyfikowano, opisywano ludzi z czterdziestoma czterema chromosomami, którzy mieli dwa chromosomy z translokacją robertsonowską. Było to w wyniku krzyżowania się dwóch osobników z kariotypem czterdzieści pięć. I ci nosiciele translokacji robertsonowskich mają problemy z niepłodnością, natomiast u tych osobników, u których nastąpiła redukcja liczby chromosomów z czterdziestu sześciu do czterdziestu czterech…
K.G.: Czyli ci z czterdziestoma pięcioma mają problemy z niepłodnością.
P.S.: Tak. U osobników z liczbą czterdzieści cztery nie stwierdzono żadnych nieprawidłowości w czasie spermatogenezy.
K.G.: A potomstwo jest zdrowe?
P.S.: Tak. Ale tylko w wypadku, kiedy dochodzi do krzyżowania się jednego osobnika czterdzieści cztery i drugiego osobnika czterdzieści cztery, co bardzo trudno uzyskać. Ale teoretycznie jest to możliwe.
K.G.: Czyli człowiek, który ma czterdzieści cztery, nie może mieć potomstwa z większością ludzi, którzy mają czterdzieści sześć?
P.S.: Jest bardzo dużo poronień, tutaj wchodzimy w mejozę i segregację chromosomów. Nosiciele translokacji robertsonowskich mają duże problemy z niepłodnością.
K.G.: Nie znamy takiego przypadku, żeby osoba z czterdziestoma czterema spotkała się z drugą osobą z czterdziestoma czterema? Widzę, że pan szuka w głowie, ale chyba nie. Natomiast biologicznie byłoby to możliwe.
P.S.: Tak, jak najbardziej.
K.G.: Proszę tu wybaczyć, drodzy słuchacze, że mówimy „samiec Homo sapiens”, „osobnicy”, ale po prostu mówimy o ludziach jako o organizmach biologicznych i stąd taka techniczna nomenklatura. Rozumiem, że to nie jest tak, że ten efekt założyciela mógł się w historii wydarzyć tylko raz. On się mógł wydarzyć więcej razy, ale tylko potomkowie tego jednego przeżyli do teraz, tak? Skoro to są przypadkowe zmiany, to mogło się tak wydarzyć, tylko tamtej części mogło nie pójść.
A.G.: Chyba kluczowe jest to, co Paweł mówił wcześniej, że te robertsonowskie mają różne miejsca.
P.S.: To po pierwsze, a po drugie tam dochodzi jednak do tego połączenia w obrębie sekwencji niekodujących. Natomiast nasza fuzja jest podobna, ale jednak troszkę różna od takiej klasycznej translokacji robertsonowskiej. Spora część materiału genetycznego została utracona, w tym geny, np. gen FOXD4L, i przez to bardzo unikalne, a te chromosomy, które wciąż są u szympansa, goryla, orangutana – 2A, 2B, nie są takie klasyczne, typowe, akrocentryczne, ale bardziej submetacentryczne, czyli troszeczkę inne. W związku z tym mimo tego, że jest tam również bardzo duża koncentracja tych segmentalnych duplikacji, bardzo trudno sobie wyobrazić, żeby to było…
K.G.: Proszę mi wybaczyć i proszę mnie zrozumieć, ale zawsze biologowie, genetycy mówili, że różne rzeczy się dzieją to tu, to tam, że to nie jest tak, że tutaj wydarzyła się jedna rzecz. A państwo przychodzą i mówią rzecz trochę wywracającą mi tę wiedzę do góry nogami. Kiwają państwo głowami z zadowoleniem. [śmiech]
P.S.: Nasz genom jest bardzo dynamiczny. Rearanżacje genomowe, aberracje chromosomowe wciąż zachodzą. I my o tym wiemy. Najczęściej w komórkach rozrodczych w czasie mejozy. Tak że ta ewolucja naszego genomu ciągle postępuje. My wciąż ewoluujemy. Widzimy to np. poprzez powstawanie różnych chorób genomowych.
K.G.: Czyli ewolucja jest cały czas?
P.S.: Cały czas.
K.G.: Bo wie pan, niektórzy mówią, że już nie ma, bo medycyna pozwala żyć tym, którzy by we wcześniejszych czasach nie przeżywali, nie mieli potomstwa, więc my zablokowaliśmy ewolucję.
P.S.: Ewolucja ciągle postępuje. Oprócz tego, że nasz genom jest taki niestabilny i kruchy, jest jednocześnie bardzo zmienny.
K.G.: Tak samo jak widać wady i zalety.
P.S.: Tak jest.
K.G.: Wady pewnie dla jednostek, ale zaleta może być dla całej populacji czy gatunku. Pani profesor, chciałabym panią wypytać jeszcze o te wątki matematyczne. Czy kiedy pani zasiada przed komputerem i modeluje lub pisze program, jeden parametr, jedna rzecz może spowodować, że się pani pomyli np. o te dwieście, trzysta tysięcy lat? Na ile one są wrażliwe na takie trochę decyzje badaczy? Jakieś założenia czy jakość danych. Jak to wygląda?
A.G.: Trzeba odróżnić od metod obliczeniowych… Moje pierwsze projekty dotyczyły identyfikacji wspomnianych elementów transpozonowych.
K.G.: A czym są transpozony?
A.G.: To są takie elementy DNA, które pochodzą z jakiegoś innego organizmu. Możemy wprowadzić całą klasyfikację transpozonów, dzielić na rodziny. Te same rodziny występują w roślinach, grzybach, ludziach, rybach. To jest zupełnie niesamowity świat. Mój przyjaciel odkrywał nowe transpozony, nazwał je transpozony Krak, bo pochodzi z Krakowa. Pomagałam mu pisać takie programy, które identyfikują. Więc trochę jest inaczej, jak to jest program, który analizuje pewne dane i ma coś znaleźć. Wtedy nie grozi pomyłka w szacowaniach i predykcjach. Oczywiście trzeba bardzo dobrze sprawdzić, czy nie ma żadnej pluskwy w programie, jak to mówią programiści. Jeśli tzw. Big Data automatycznie się analizuje, zawsze jest ryzyko, że jakaś pluskwa się wkradnie i nie będzie się dało potwierdzić hipotezy, którą wygenerujemy. Jak zastanawiamy się nad jakimś modelem matematycznym, to zazwyczaj narzędziem, które stosujemy, jest domieszka rachunku prawdopodobieństwa. Zakładamy, że to, co się dzieje, zachodzi z pewnym prawdopodobieństwem i często te modele są probabilistyczne. Co oznacza, że jesteśmy w stanie skwantyfikować to ryzyko. Możemy sprawdzić, jak dobrze nasze modele przewidują. Jeżeli robimy coś na danych z eksperymentów molekularnych i nie znamy odpowiedzi na nasze pytanie, to nie możemy powiedzieć, czy dobrze przewidzieliśmy tę odpowiedź. Ale możemy sobie samemu wygenerować sztuczne dane z naszego modelu, dla których znamy odpowiedzi, i sprawdzić, czy model rzeczywiście tę odpowiedź zwraca. Testujemy. Zanim się pochwalimy w artykule, że coś oszacowaliśmy, staramy się przetestować, czy możemy mieć zaufanie do naszej metody.
K.G.: Ale mówiła pani też o tym błędzie. Rozumiem, że jest pani w stanie powiedzieć trochę jak socjologowie, że ten nasz wynik może być przesunięty o ileś tam do przodu i ileś tam do tyłu.
A.G.: Tak, że jest obarczony takim błędem. Najbardziej prawdopodobny wynik jest taki, ale jest możliwość.
K.G.: Jak państwo zaczęli ze sobą współpracować i ile państwu zajęło dojście do tego wniosku?
P.S.: Zaczęliśmy piętnaście lat temu. Jeszcze zanim się rozwinęły techniki sekwencjonowania nowej generacji, dostaliśmy wspólny grant z NCBiR-u na wprowadzenie do diagnostyki klinicznej tzw. mikromacierzy. Pracowaliśmy nad tym wspólnie i była to współpraca między Uniwersytetem Warszawskim, Baylor College of Medicine oraz Instytutem Matki i Dziecka, Zakładem Genetyki Medycznej, którego kierownikiem był wówczas pan profesor Tadeusz Mazurczak. I dzięki naszej współpracy udało nam się skonstruować własną tzw. macierz, a potem ją zaimplementować i wprowadzić do rutynowej diagnostyki genetycznej, klinicznej, która do tej pory jest używana. Uważa się nawet, że do identyfikacji zmienności liczby kopii DNA jest bardziej czułą metodą niż techniki sekwencjonowania nowej generacji.
A.G.: I została wprowadzona w Instytucie Matki i Dziecka do diagnostyki wiele lat temu.
K.G.: Z tego, co państwo mówią, jesteście przykładem tego, o czym się w nauce, mówiąc brzydko, trąbi, ale nie zawsze łatwo jest to wprowadzić w życie, czyli właśnie tej interdyscyplinarności. Pani się uśmiecha.
A.G.: Tak, ona jest odmieniana przez wszystkie przypadki, jest w dobrym tonie mówić, że interdyscyplinarność jest ważna i projekty interdyscyplinarne powinny być finansowane. Niestety, jak dochodzi co do czego, to okazuje się, że jest to trudne i wymaga nie tylko poznania obcej dziedziny wiedzy…
K.G.: Pani się uczyła genetyki?
A.G.: Musiałam nauczyć się zarówno genetyki, jak i technologii, biotechnologii. Żeby stworzyć jakąś metodę, która analizuje dane z nowoczesnej technologii, trzeba tę technologię poznać, jest to konieczne. Bo właściwie mówimy, że współczesna biologia i medycyna molekularna jest napędzana przez rozwój technologii. A ten rozwój technologii musi iść od razu w parze z rozwojem nowych metod analizy, modeli matematycznych, metod informatycznych. Bo to nie są technologie, które można ręcznie przeanalizować. To są gigabajty danych.
K.G.: Z kolei pan uczył się informatyki?
P.S.: Troszkę się uczyłem. Ale rzeczywiście, zgodzę się, że kluczem naszej współpracy był wspólny język, tak, żebyśmy poznali wzajemnie swoje dziedziny i wtedy nastąpił taki efekt synergistyczny, że zaczęło to świetnie działać.
K.G.: Chciałam pana jeszcze wypytać o wspomnianego na początku krótko profesora Jamesa Lupskiego, który zauważył, że te wieloliterowe, wielozdaniowe zmiany, jak mówiliśmy, porównując nasz genom do książki, mogą być odpowiedzialne za konkretne choroby. Pan trochę trzymał za niego kciuki, żeby zdobył w tym roku Nobla.
P.S.: Wciąż trzymam. Miałem ten zaszczyt i przyjemność współpracować z profesorem Lupskim od 2000 roku w Baylor College of Medicine. Kiedy tam przyjechałem w latach dziewięćdziesiątych, profesor Lupski użył tego określenia „choroby genomowe”.
K.G.: To dawno, przed pierwszym sekwencjonowaniem.
P.S.: Tak, to były początki. Zajął się troszkę tym tematem w związku ze swoją chorobą, ponieważ ma on obwodową polineuropatię, która jest uwarunkowana różnymi genami. W 1991 roku stwierdził, że najczęstsza postać tej choroby spowodowana jest zmianą liczby kopii. Mianowicie trzecią kopią genu BNP22 na chromosomie siedemnastym. I potwierdził to różnymi metodami molekularnymi. Bardzo trudno było się przebić w środowisku z taką zupełnie rewolucyjną zmianą, kiedy jeszcze genom człowieka nie był sekwencjonowany, że to nie mutacja pojedynczego nukleotydu, ale zmiana kopii jednego genu może powodować zmiany i może być tak częstą przyczyną tej choroby. Okazało się, że akurat nie ta najczęstsza postać jest przyczyną jego choroby. Dopiero po wielu latach, kiedy zastosowano u niego sekwencjonowanie eksonu, genomu, udało się znaleźć gen, który jest zmutowany w jego przypadku. Jest to postać recesywna tej choroby. On całe swoje życie od dzieciństwa miał kontakt ze szpitalami, przeszedł wiele operacji ortopedycznych. Tak się tym zainteresował, że skończył medycynę w Nowym Jorku, zrobił doktorat i przeniósł się stamtąd do Teksasu, do Houston, do Baylor College of Medicine, gdzie zrobił rezydenturę z pediatrii, z genetyki klinicznej i jednocześnie był tam lekarzem klinicystą i genetykiem, który kontynuował swoje badania naukowe w laboratorium. Do tej pory je kontynuuje. Ma ogromne osiągnięcia, wiele publikacji, wiele odkryć. Jest pionierem, liderem w tej dziedzinie i takim człowiekiem renesansu w genetyce, który rozwinął wiele kierunków i to wszystko jakoś ogarnia.
K.G.: Oczywiście miało to przełożenie na życie pacjentów, bo zaczęli być diagnozowani. Kolejne jednostki chorobowe zaczęły być nazywane, zaczęto znajdować ich przyczyny?
P.S.: Tak. Oczywiście nie tylko obwodowa polineuropatia, ale dzięki wprowadzeniu metod do genomowej analizy, czyli najpierw tych mikromacierzy, a potem sekwencjonowania eksonu, potem genomu, mamy techniki i metody, które zrewolucjonizowały medycynę w każdej jej dziedzinie. Diagnostyka kliniczna, genetyczna oczywiście umożliwia stawianie precyzyjnych diagnoz molekularnych i pomaga diagnozować choroby w rodzinach, które przez wiele lat tułały się od jednego lekarza do drugiego, przechodziły różne badania, tomografy. Dopiero dzięki badaniom genetycznym można to jednoznacznie precyzyjnie zidentyfikować.
K.G.: Czy opowie nam pan trochę więcej o tym, co się dzieje w Texas Medical Center? Bo jak zajrzałam tam do statystyk, to można się za głowę złapać. Przeogromne centrum medyczne – dziesięć milionów pacjentów rocznie. To się w głowie nie mieści.
P.S.: Baylor College of Medicine jest prywatną wyższą szkołą medyczną, która rozwinęła się po II wojnie światowej. Jest zlokalizowana w samym centrum tzw. Texas Medical Center, które jest uważane za największe centrum medyczne na świecie, w którym pracuje codziennie sto dwadzieścia tysięcy ludzi. Jest tam wiele znanych najlepszych szpitali w Stanach Zjednoczonych. Również na świecie, np. szpital onkologiczny MD Anderson Cancer Center, który jest jednym z dwóch najlepszych w Stanach. Albo Texas Children’s Hospital – jeden z trzech najlepszych szpitali dziecięcych. Oraz wiele innych szpitali i instytucji medycznych. Natomiast Baylor College of Medicine ma bardzo rozbudowaną część naukową z badaniami naukowymi i wiele zakładów, wiele jego specjalności jest przodującymi w Stanach i uważa się je za najlepsze tamtejsze zakłady, w tym właśnie nasz Zakład Genetyki Molekularnej i Genetyki Człowieka, który pod względem liczby grantów, finansowania czy jakości badań, publikacji od wielu, wielu lat jest liderem i jest uważany za numer jeden w Stanach, a przez to pewnie na świecie.
K.G.: Proszę wybaczyć, jak to wygląda? Czy jest to taki odpowiednik Las Vegas, tylko medyczny, że nagle na pustyni wyrastają szpital obok szpitala? Jak to wygląda?
P.S.: Tak, Houston w ogóle jest czwartym miastem w Stanach pod względem liczby ludności. Natomiast Texas Medical Center jest jakby miastem w mieście. Te wszystkie budynki są zlokalizowane w jednym miejscu i świetnie to funkcjonuje. To centrum wciąż się rozrasta. Pamiętam recesję z lat 2008-2009 – nie widziałem tam żadnego śladu recesji, tam wciąż się to rozrastało.
K.G.: Na zdrowiu nikt nie oszczędza, przynajmniej jeśli chodzi o pacjentów.
P.S.: Ten system finansowania bardzo sprawnie tam funkcjonuje. Mimo że można mieć zastrzeżenia, bo niestety ubezpieczenia w Stanach nie są dla wszystkich. Houston jest uważane za centrum energetyczne świata, gdzie są podpisywane wszystkie kontrakty związane z ropą naftową, a Texas Medical Center jest drugim źródłem dla miasta.
K.G.: Natomiast to nie jest tak, że współpraca z Uniwersytetem Warszawskim była uprzejmościowa, bo wiem, że informatyka jest tam znakomita, jest absolutnie na światowym poziomie.
P.S.: O czym Amerykanie dobrze wiedzą.
A.G.: Można powiedzieć, że nasza informatyka jest na światowym poziomie. Jak popatrzymy na takie prestiżowe granty Europejskiej Rady Nauki, które są przyznawane naprawdę na wyjątkowe projekty, to w informatyce my zazwyczaj mamy ich tyle, co Oxford i Cambridge razem wzięte. Ta informatyka uprawiana w naszym instytucie naprawdę jest na najwyższym światowym poziomie. Oczywiście jest to bardziej informatyka koncentrująca się na metodach bliskich matematyce, bo nasz wydział jest wydziałem matematyki i informatyki. Wspólnie, co się nie tak często zdarza. I my rzeczywiście czerpiemy, jest to taki efekt synergii. Współpracujemy między sobą i kultura matematyczna naszych informatyków jest ogromna, co umożliwia im świetne działanie w takich dyscyplinach jak kryptografia, algorytmika, logika. To bardzo dużo daje.
P.S.: Jeszcze chciałbym dodać, że mamy bardzo owocną współpracę z Politechniką Warszawską, z bratem Ani – panem profesorem Tomaszem Gambinem. Mamy wiele wspólnych publikacji. I ta wymiana, nasza naukowa współpraca czy to z genetykami z Instytutu Matki i Dziecka, czy właśnie z informatykami z uniwersytetu, czy z politechniki, zaowocowała wieloma doktoratami, habilitacjami, również profesurami. Tak że bardzo, bardzo owocna współpraca.
K.G.: Udaje się pani namawiać kolegów, koleżanki, doktorantów, doktorantki, żeby się tej genetyki uczyć?
A.G.: Tak, bez żadnego problemu. Do tej pory wypromowałam jedenastu doktorantów, z czego pięciu w tematyce genetyki. We współpracy z Pawłem wypromowaliśmy razem Piotra. Praktycznie wszyscy moi doktoranci wizytowali Baylor College of Medicine, odwiedzali Pawła.
P.S.: W zasadzie każdy taki pobyt dwu-, trzymiesięczny kończył się przynajmniej jedną publikacją. Tak że wszyscy byli z tego zadowoleni.
K.G.: Czyli przekładało się to na konkrety. Szanowni państwo, co dalej? Jakie dalsze plany badawcze? Coś państwo zdradzą? Ludzie są zafascynowani tym tematem. W Radiu Naukowym odcinki dotyczące pochodzenia człowieka są bardzo popularne. Marzymy o tym, żeby się tego wreszcie dowiedzieć, żeby ten łańcuszek jakoś uzupełnić. Jakie plany?
A.G.: Chcemy się trochę zająć ewolucją molekularną, ale ewolucją regulacji.
K.G.: Czyli tej gąsienicy i motyla.
A.G.: Tak. Jak w czasie ewolucji genetycznej postępowała ewolucja tej sieci interakcji genów. Zamierzamy tutaj użyć teraz będących na topie metod deep learningu, czyli głębokiego uczenia, uczenia maszynowego, i próbować odtworzyć, jak zmiany genetyczne przenoszą się na zmiany regulacyjne.
P.S.: A bardziej konkretnie właśnie dostałem grant na następne cztery lata z National Institute of Health na badanie genetyki, genomiki, śmiertelnych chorób rozwojowych płuc u noworodków. Moje laboratorium odkryło kilka krytycznych genów w rozwoju płuca i stwierdziliśmy, że w rozwoju, ale prawdopodobnie również w ewolucyjnym powstaniu płuca kluczową rolę odegrały sekwencje regulatorowe, nie tylko te kodujące.
A.G.: Ten temat zbliża się też troszkę do ewolucji, ponieważ badanie tego, jak rozwijają się płuca w rozwoju płodowym, trochę nas zbliża do badania tego, jak płuca wyewoluowały ze skrzeli w momencie, kiedy ryby wychodziły na ląd. Mamy już jedną rybkę zsekwencjonowaną i też tam moja doktorantka zaczęła badać, jakie są obszary regulatorowe.
K.G.: Jak się państwu to wszystko mieści w głowie? Chciałam się podzielić – raz na jakiś czas mam taką impresję, nie chcę powiedzieć kreacjonistyczną, ale myślę, że stąd wynika opór części osób, że przecież te płuca są skomplikowane, to oko jest skomplikowane, że gdzie to mogło powstać z przypadkowych mutacji, z przypadkowych zmian. Czy państwo raczej pracują na takich kawałeczkach, takich małych puzzlach i wtedy się wszystko zgadza? Ale jak spojrzycie tak z góry, to nie łapiecie się czasami za głowy, że to jest po prostu nieprawdopodobne, że do tego doszło? A jest. Macie jakieś emocje z tym związane?
A.G.: Z drugiej strony sama pani wspomniała, że fuzja była tylko raz, więc możemy powiedzieć, że to było takie nasze wyjście z raju, jednokrotne. Wybraliśmy siebie jako ludzi, natomiast straciliśmy bycie w jedności z całą inną przyrodą. To prawda, że jest to niesamowite, ale właśnie dlatego tak bardzo fascynujące. Przynajmniej poznawczo to nie jest żadna przeszkoda, że coś jest skomplikowane i nie widzę tutaj konieczności wprowadzania jakichś upraszczających schematów myślowych. Dla mnie jest oczywiste, że jeśli przyjmiemy koncepcję istnienia Boga, to Pan Bóg stworzył ewolucję. I zrobił to najlepiej, jak mógł.
K.G.: Rozumiem te odniesienia do religii, bo one są naturalne i części z naszych słuchaczy też na pewno to przychodziło do głowy, np. z tym momentem, że raz się wydarzyło. Moja emocja raczej jest taka, że właśnie to nieprawdopodobnie skomplikowane, że ja tu do państwa mówię, ktoś wymyślił tę technologię, żebyśmy później byli słyszani. A to gdzieś tam jakieś białko zahacza o siebie i w ogóle to się źle skopiowało – wiecie, jak schodzimy do takiego poziomu, a produkt jest taki, jakim jesteśmy my czy inne fenomenalne organizmy, no to czasami właśnie jest taki moment, że wow, jak to? Jak to jest możliwe?
A.G.: To jest potęga tej ewolucji.
P.S.: Dokładnie. Szczęśliwie żyjemy w takim czasie, gdzie mamy nowe techniki, metody, które działają, którymi możemy badać, mamy na to finansowanie. Temat jest fascynujący, ekscytujący, który naprawdę motywuje, zachęca, napędza nas do pracy. No i to pokazuje, że warto inwestować w naukę, w badania naukowe, bo to przynosi naprawdę wielokrotny zysk. Sam tego doświadczyłem, wielokrotnie to widziałem. Mimo że badania naukowe czasami są frustrujące, czeka się wiele tygodni, miesięcy, czasami lat. Ale warto. Przykładem jest odkrycie tego mechanizmu fuzji, nad którym pracowaliśmy pięć, sześć lat.
A.G.: Tak, to jest ogromna zasługa wspomnianej już Basi Poszewieckiej, która nie zniechęcała się, układała te puzzle, co naprawdę nie było łatwe, bo były ogromne obszary błękitnego nieba.
P.S.: Ważne, żeby się nie poddawać.
K.G.: Dlatego trzeba też młodych w nauce, bo entuzjazm również jest potrzebny. Pani profesor Anna Gambin i pan profesor Paweł Stankiewicz. Dziękuję państwu serdecznie za rozmowę.
P.S.: Bardzo dziękujemy.
A.G.: Dziękujemy pięknie.
Udostępnij:
Pracuje na Wydziale Matematyki, Informatyki i Mechaniki Uniwersytetu Warszawskiego. Specjalizuje się w algorytmach biologii obliczeniowej, algorytmach randomizacyjnych oraz obliczeniach odpornych na błędy.
Profesor w Baylor College of Medicine w Houston, Teksas, USA. Jego badania skupiają się na lepszym zrozumieniu mechanizmów molekularnych oraz konsekwencji fenotypowych rearanżacji genomu








